







实验旨在了解RNA-seq的基本原理。通过模仿文献《Targeting super enhancer associated oncogenes in oesophageal squamous cell carcinoma》的流程,学会利用NCBI和EBI数据库下载数据,熟悉Linux下的基本操作,并使用R语言画图,用Python或者shell写脚本进行基本的数据处理,使用FastQC等软件进行数据处理,学习Hisat2+StringTie+Ballgown的RNA-seq分析流程,并对结果进行分析讨论。
1、硬件平台
处理器:Intel(R) Core(TM)i7-4710MQ CPU @ 2.50GHz
安装内存(RAM):16.0GB
2、系统平台
Windows 8.1,Ubuntu
3、软件平台
① Aspera connect ② FastQC ③ Hisat2
④ StringTie ⑤ Ballgown
4、数据库资源
NCBI数据库:https://www.ncbi.nlm.nih.gov/;
EBI数据库:http://www.ebi.ac.uk/;
5、研究对象
加入H3K27Ac 抗体处理过的TE7细胞系测序数据和其空白对照组
加入H3K27Ac 抗体处理过的KYSE510细胞系和其空白对照组
背景简介:食管鳞状细胞癌(OSCC)是一种侵袭性的恶性肿瘤,本文章通过高通量小分子抑制剂进行筛选,发现了一个高度有效的抗癌物,特异性的CDK7抑制剂THZ1。RNA-Seq显示,低剂量THZ1会对一些致癌基因的产生选择性抑制作用,而且,对这些THZ1敏感的基因组功能的进一步表征表明他们经常与超级增强子结合(SE)。Chip-seq解读在OSCC细胞中,CDK7的抑制作用的机制。
本文亮点:确定了在OSCC细胞中SE的位置,以及识别出许多SE有关的调节元件;并且发现小分子THZ1特异性抑制SE有关的转录,显示强大的抗癌性。
文章PMID: 27196599
1、Aspera软件下载及安装
进入Aspera官网的Downloads界面,选中aspera connect server,点击Wwindows图标,选择v3.6.2版本,点击Download进行下载。
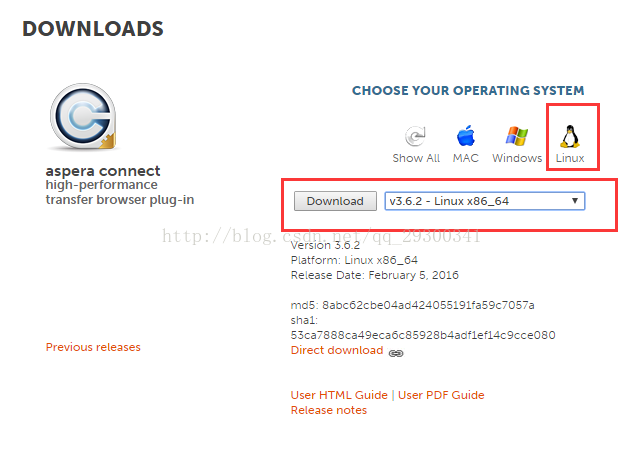
图表 1 aspera的下载
Linux下的安装配置参考博文:
http://blog.csdn.net/likelet/article/details/8226368
2、RNA-Seq数据下载
1)选择NCBI的GEO DataSets数据库,输入GSE76861,打开GSM2039114、GSM2039119、GSM2039120、GSM2039125获取它们对应的SRX序列号。(此处仅仅做了KYSE510和TE7细胞系加入THZ1后0hr和12hr的RNA-seq数据)
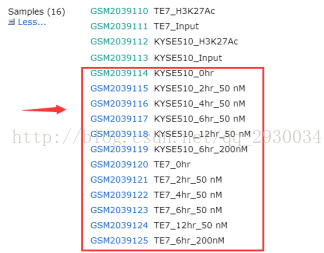
图表 2 RNA-seq数据

图表 3 获取SRA编号
2)进入EBI,获取ascp下载地址
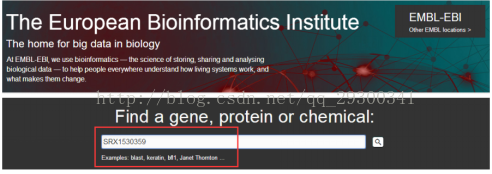
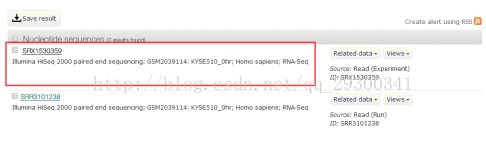

图表 4 ascp下载地址
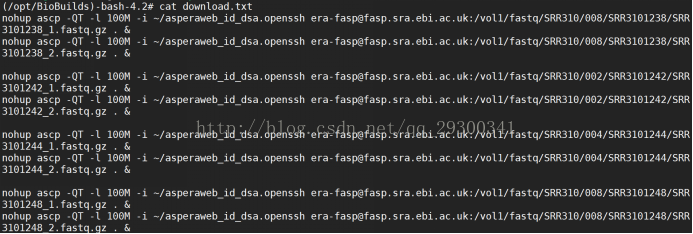
3)使用aspera下载并解压
aspera下载命令及gunzip解压命令(nohup+命令+& 可以后台运行)
3、FastQC质量检查
3.1 FastQC的安装
Ubuntu软件包内自带Fastqc
故安装命令apt-get install fastqc
3.2使用FastQC进行质量检查
fastqc命令:
fastqc -o . -t 5 SRR3101238_1.fastq.gz &
-o . 将结果输出到当前目录
-t 5 表示开5个线程运行
(要分别对八个fastq文件执行八次)
4、使用Hisat2对Reads进行Mapping
4.1 Hisat2的安装
进入Hisat2官网http://ccb.jhu.edu/software/hisat2/index.shtml下载二进制程序,再配置环境变量即可。
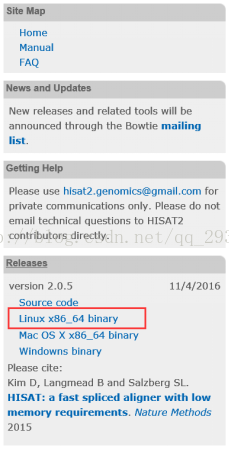
图表 5 Hisat2下载与安装
4.2下载人类参考基因组和注释文件
人类参考基因组:Hisat2官网上有Ensemble GRCh38的基因组索引,可以直接下载使用
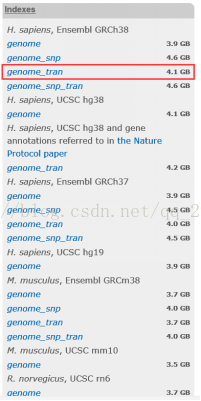
图表 6 Hisat2建立的GRCh38索引
注释文件:下载自ensemble数据库ftp://ftp.ensembl.org/pub/release-86/gtf/homo_sapiens
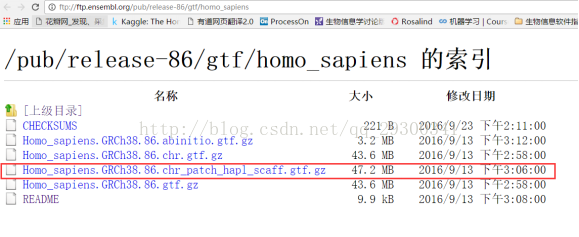
图表 7 Ensemble注释文件
PS:如果是想自己下载基因组,然后自己建立索引,需要使用Hisat2包里面的python脚本
extract_splice_sites.py和extract_exons.py,从注释文件里面抽取出剪切位点和外显子信息
First, using the python scripts included in the HISAT2 package, extract splice-site and exon information from the gene annotation file:
extract_splice_sites.py chrX_data/genes/chrX.gtf >chrX.ss
extract_exons.py chrX_data/genes/chrX.gtf >chrX.exon
Second, build a HISAT2 index:
hisat2-build --ss chrX.ss --exon chrX.exon chrX_data/genome/chrX.fa chrX_tran
The --ss and --exon options can be omitted in the command above if annotation is not available.
4.3 Hisat2比对
将RNA-seq的测序reads使用hisat2比对
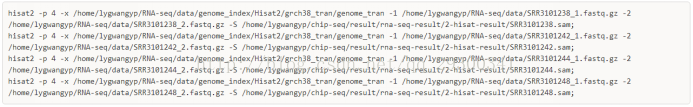
5、samtools文件格式转化
samtools将sam文件转成bam,并且排序,为下游分析做准备
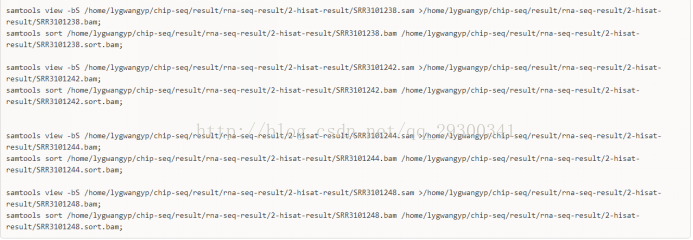
6、stringtie转录本处理
6.1stringtie组装转录本
stringtie对每个样本进行转录本组装
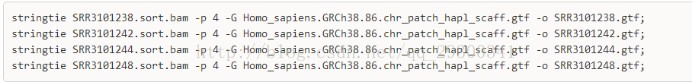
6.2stringtie合并转录本
stringtie 将所有样本的转录本进行合并
注意:此处的mergelist.txt是自己创建的,需要包含之前SRR3101238.gtf,SRR3101242.gtf,SRR3101244.gtf,SRR3101248.gtf的路径
stringtie --merge -p 4 -G Homo_sapiens.GRCh38.86.chr_patch_hapl_scaff.gtf -o stringtie_merged.gtf mergelist.txt;
6.3 stringtie评估表达量
计算表达量并且为Ballgown包提供输入文件
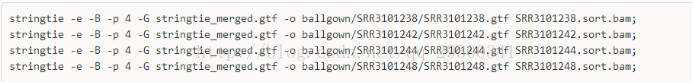
7、Ballgown表达量分析
7.1 Ballgown的安装
source("http://bioconductor.org/biocLite.R")
biocLite("ballgown")
7.2 文件准备与分析
将数据的分组信息写入一个csv文件,此处phenodata.csv文件
phenodata.csv文件内容
sampleid,celllines,time
SRR3101238,KYSE,0hr
SRR3101242,KYSE,12hr
SRR3101244,TE7,0hr
SRR3101248,TE7,12hr
接下来就可以用ballgown愉快地进行分析了。
此处只做了四个样本,然后在Ballgown包里用stattest是无法计算出P值的,样本稍微多一些才可以检验。
1、RNA-Seq数据下载
RNA-Seq数据下载结果
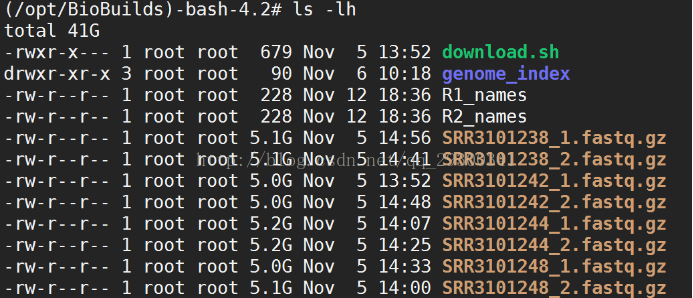
图表 8 RNA-Seq数据
2、FastQC质量检查
数据质量检查
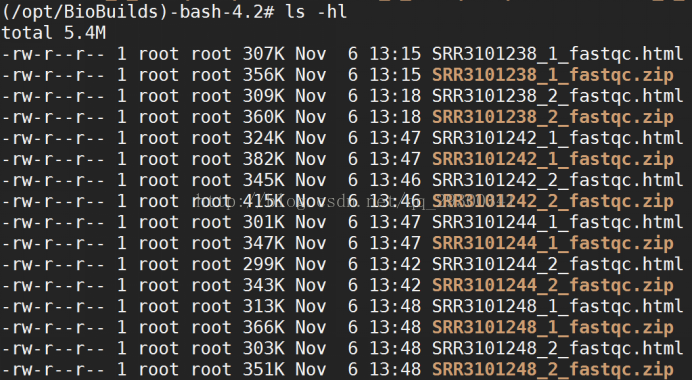
图表 9 质量检查文件
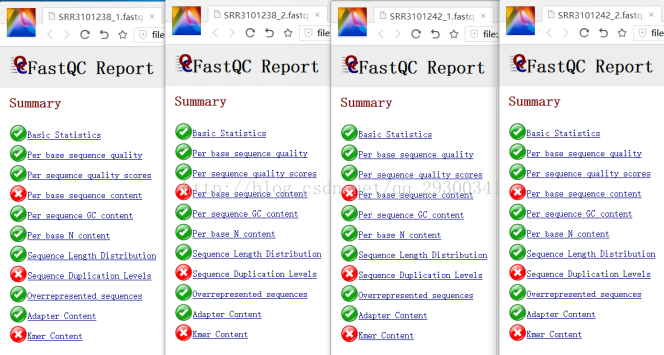
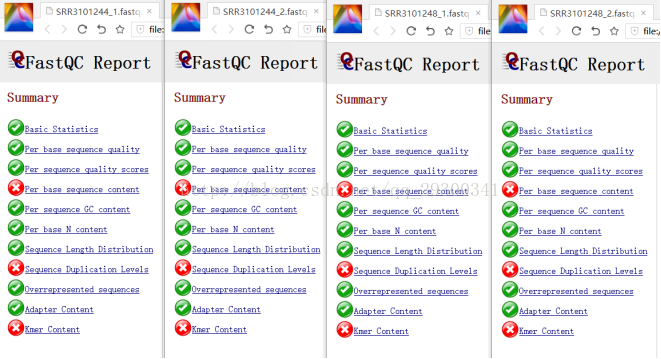
3、使用Hisat2对Reads进行Mapping
3.1基因组文件
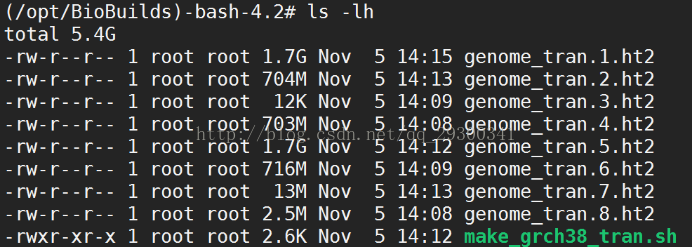
图表 11人类参考基因组GRCh38索引
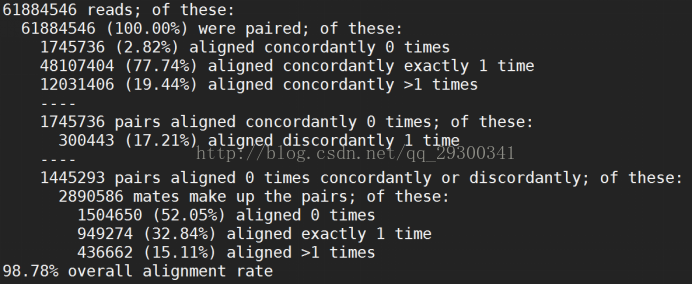
3.2 Mapping结果
4、samtools结果
samtools结果文件
图表 13文件转化结果
5、stringtie结果
5.1组装转录本
图表 14 组装转录本
5.2 合并转录本
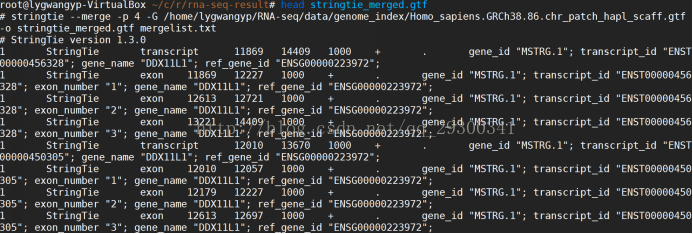
图表 15合并转录本
5.3评估表达量
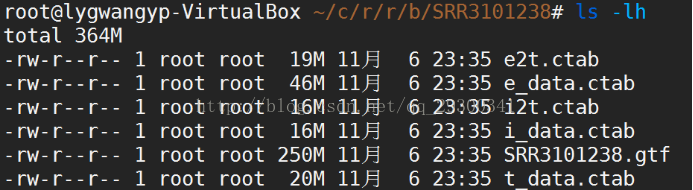
图表 16评估表达量
6、Ballgown分析结果
剪切方式分析
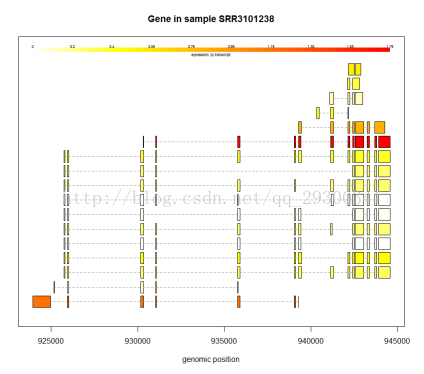
图表 17 某个基因的剪切方式
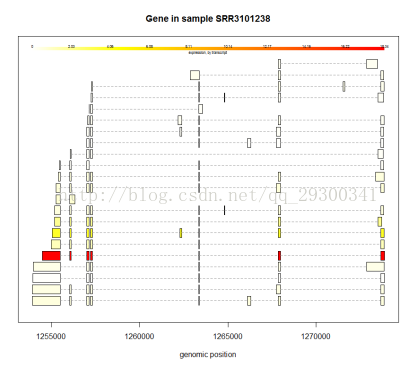
图表 18某个基因的剪切方式
FastQC质量检查
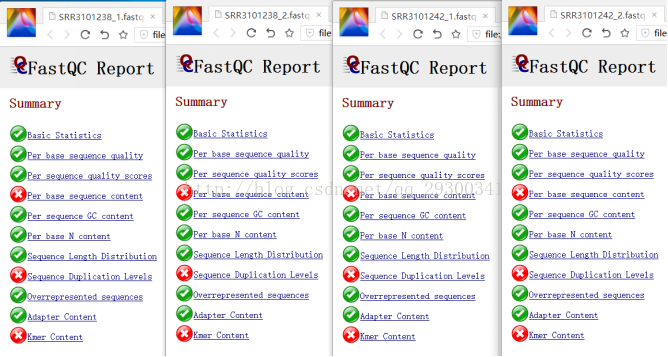
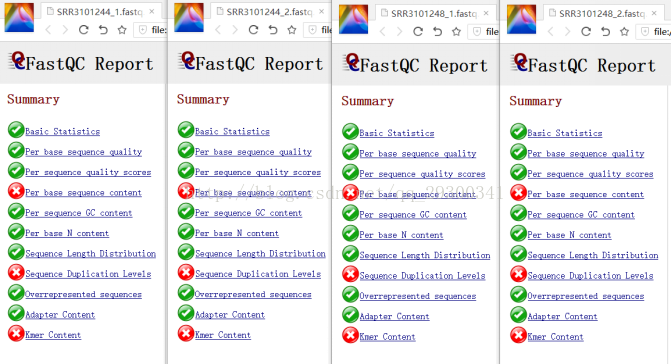
图表 10 质量检查结果显示,测序质量挺好,Per base sequence content、Sequence Duplication Levels、Kmer Content出现警告更可能是由于测序方法本身存在的固有误差。
Hisat2整体覆盖度
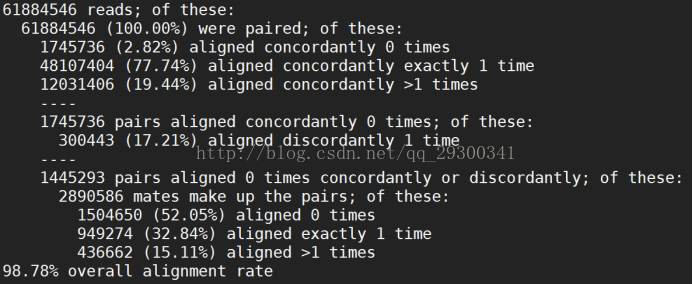
由图表 12 Mapping整体结果可以看出,四个fastq文件Mapping整体覆盖率都在98%左右,从另一方面说明数据质量很好
可变剪切方式分析
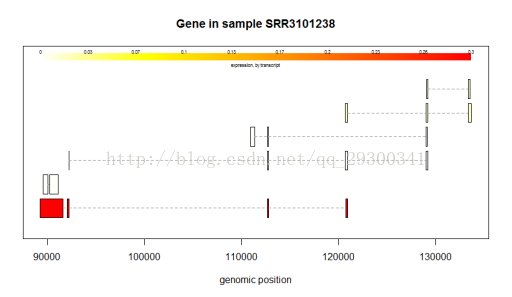
初步绘制了几个基因的剪切方式图,发现在可变剪切中,确实会有某一种方式占据主导地位。
表达量热力图
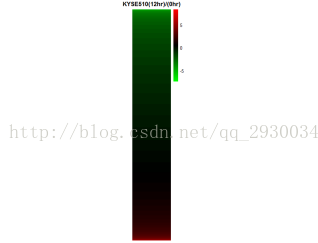

图表 19 KYSE510细胞系和图表 20 TE7细胞系的基因表达图的计算方式为:
加药12hr表达量÷0hr表达量,再取log2后的值;
由于表达量信号值小于1不显著,故筛去,仅留下信号值都大于1的基因。
从图中绿色较多,可以看出加入THZ1的12hr后,明显抑制了基因的表达。
RNA-Seq与Chip-Seq结合分析
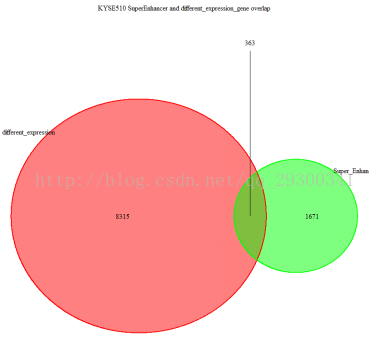
图表 21 KYSE510结合分析显示,KYSE510细胞系差异表达基因中,有363个与Chip-seq分析流程中鉴定出的SuperEnhancer相重叠。
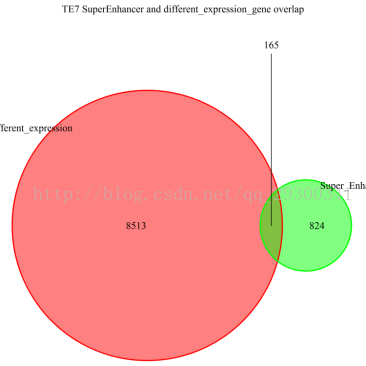
图表 22 TE7结合分析显示,TE7细胞系差异表达基因中,有165个与Chip-seq分析流程中鉴定出的SuperEnhancer相重叠。
Kobas注释分析
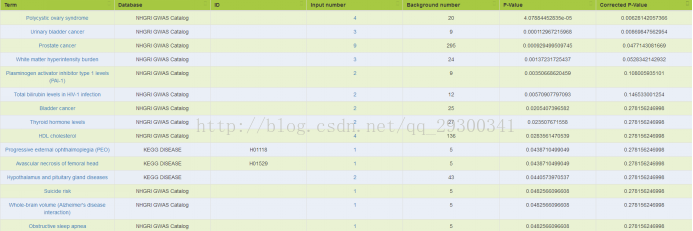
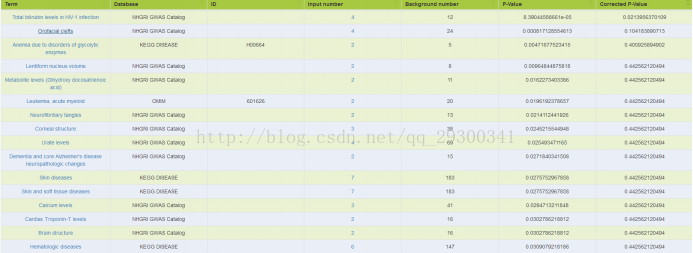
对于Chip-seq中SuperEnhancer和RNA-seq差异表达基因overlap的部分,先用Ensemble数据库的Biomart,将GeneSymbol转成EnsembleID,再输入至Kobas注释,注释选用OMIM、KEGG DISEASE、NHGRI GWAS Catalog数据库,由图表 23 TE7结合分析基因注释和图表 24 KYSE510结合分析基因注释结果来看,与癌症相关。
参考文献
[1] Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma PMID: 27196599














 292
292











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









