







3 基于Hadoop基因测序数据处理关键技术的研究
3.1 测序处理流程与MapReduce结合
基因测序处理流程中有gVCF和VCF文件生成两个阶段,而MapReduce恰好是一种分阶段处理的编程模型,它拥有Mapper和Reducer两个处理阶段,因此将MapReduce框架和测序流程结合在一起,既能保证测序流程的有序进行,又能在MapReduce框架中进行并行调度,提升测序数据处理的效率。
3.1.1 测序流程与MapReduce结合的可行性分析
MapReduce是一种可用于数据处理的编程模型,可以将数据分析处理流程分为映射阶段和归约阶段1。在映射阶段,各个Map程序在不同的Container中运行彼此互不干扰,即各个Mapper阶段的程序本质上是并行运行的;而到了Reducer规约阶段,将各个Mapper阶段产生的中间结果做规约整合得到最终的处理结果。因此可以将大规模的数据分析任务分发给任何一个拥有足够多机器的数据中心,也正是因为其强劲的数据分析和并行处理能力,所以选择其和测序处理流程作整合。
在原本传统的基因测序流程中,是将多个样本放在一台高性能服务器中,通过编写的测序分析脚本串行处理每一个样本数据,最终得到变异检测的结果。虽然这个过程能得到最终的结果,但需要配置高性能服务器,而且往往需要耗费大量的处理时间,这其中大部分时间花在测序比对上面。后期要想提升测序效率的话,需要进一步配置性能更高的服务器,这样一个高时间花费和昂贵开销,不利于基因测序的低成本普及和个性化医疗的推广应用。
因此,我们需要一种高效并行的处理平台,达到使原本串行处理多个样本的测序过程,能够以一个样本为测序处理单位细分为多个Map任务达到并行处理分析,再通过Reduce任务的到最终的结果。
3.1.2 测序流程与MapReduce框架的结合
要想将测序处理流程与MapReduce框架结合,首先要将对多个样本的测序切分为多个Map任务,在运行MapReduce任务的时候,将基因测序的各个流程封装在shell脚本中,而这个shell脚本是以脚本模版的形式呈现。另一方面,主要的测序分析流程是固定的,而只有分析样本对象的信息是动态传入的。这样可以通过编写一个特定的Map程序,再通过调用MapReduce驱动程序来运行作业,对于传入的不同样本名生成不同的Map任务,即各个基因测序脚本,达到基因测序的并行化处理。
在得到以样本为单位的单变异检测的文件后,通过一个Reduce任务合并所有的gVCF变异文件,从而得到最总对于整个样本数据的变异检测VCF文件。
一个集群中有三台机器,总共有九个样本数据,那么可以并行三次可以分析完所有的数据,但是若是通过传统的流程分析过程的话,需要串行分析九次才能完成整体的测序处理流程。因此,通过流程处理逻辑的MapReduce化,可以成倍的提升样本测序的处理效率,使基因测序更加高效。
3.2 不同格式数据的访问与存储
3.2.1 数据访问于存储面临的问题
在进行MapReduce处理程序时,通过shell脚本的方式处理数据,由于不同样本的处理流程需要拉取不同的数据源,并且输出的中间结果也是不同的。这个时候,我们需要一个统一的数据存储和访问规则来统一管理这些数据源以及中间结果等。
3.2.2 数据的存储与访问分析
首先,对于参考序列这一部分数据源,因为其涉及的处理流程比较固定,可以事先在机器的本地环境先搭建好。而MapReuce程序是在集群中的不同机器上运行的,因此数据是在MapReduce程序中动态获取的,我们需要事先上传到HDFS上,这也是MapReduce计算框架常用来获取数据源的方式,其他方式还包括通过访问Amason S3等。
将所有的样本数据存储在HDFS上之后,通过在Map程序的输入数据中指定样本名称和存储的位置信息,可以轻松访问到指定的样本数据,从而实现不同的Map程序访问并处理不同的样本数据。而对于中间其他剩余几个测序流程,如排序、标记重复、创建索引以及变异检测这几步,上一步的结果是下一步处理流程的输入数据,这些处理流程所得到的中间数据都可以本地化存储,既能节省网络传输的开销,也能最大程度保证数据的完整性,不会由于网络中断导致数据不完整存储。
在所有的样本并行化处理完成之后,每个Map程序最终得到的是该样本数据的gVCF变异检测文件,对每个样本变异检测数据作一个合并得到最终VCF格式文件,这时需要用到Reduce归约操作,因此需要对Map程序最终得到的结果再上传到HDFS上,供接下来的Reduce程序使用。
而最终得到的VCF文件,也是需要上传到HDFS上存储,保证结果文件的容错性和完整性。
3.3 分析流程的完整性
3.3.1 分析流程面临的问题
WGS分析处理流程中主要的问题是如何判定并以一个样本数据作为切分标准,对整体的多样本数据以样本为单位进行切分处理。因为只有以单样本为数据测序分析的单位,才能既保证数据处理流程完整性的同时,又能最大程度上保证并行化处理,从而提升整体的测序效率。
理论上对于不同样本的分析流程其实都是一样的,因此需要引入一套流程分析机制,保证分析流程的统一性,便于分析流程的统一操作和中间结果管理。
3.3.2 整体流程设计分析
在MapReduce框架中,其Mapper阶段和Reducer阶段的输入文件都是文本格式的文件,在我的整体流程搭建中,将测序的样本文件存于HDFS上,map和reduce的输入文件只是保存样本文件的列表,达到能够将不同的样本处理流程进行隔离和分批处理的目的。
具体是在Map任务的输入数据文本中,每一行指定一对R1和R2样本数据名称,在驱动程序中设置输入文件中的每一行数据用一个Mapper来处理,这样能够将每一个样本数据处理分到不同的Map任务中执行,从而达到流程设计的完整性以及流程处理的并行化。
考虑到基因测序的分析流程,像完成比对、重复标记和变异检测的大多数开源工具(如BWA、SAMtools和GATK)都提供了Linux命令行界面,所有各个MapReduce阶段中的map()和Reduce()函数将调用Linux Shell脚本,并提供适当的参数。要执行这些shell脚本,将使用FreeMarker模版语言。
3.4 FreeMarker引擎与测序流程的模版化
Apache FreeMarker2是一种模板引擎,目前是Apache基金会的一个孵化项目,模板是用FreeMarker模板语言(FTL)编写的,它是一种简单的专用语言。通常,使用通用编程语言(如Java)来准备数据(发布数据库查询,进行业务计算)。然后,FreeMarker使用模板显示准备好的数据。
在我们基因测序流程搭建中,先编写shell分析流程的脚本模板,其中分析对象用参数代替,再通过FreeMarker API将参数传入分析流程模版中,得到最终可执行的shell脚本文件。FreeMarker模版引擎如图3-1所示。
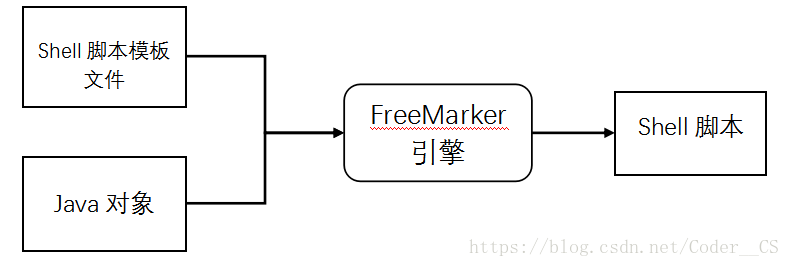
图3-1 FreeMarker模版引擎
3.4.1 构建FreeMarker模版引擎
在我们最终的处理流程中,通过FreeMarker模版引擎类把脚本模版转换为可执行的shell脚本后,最终通过调用shell脚本工具类执行该脚本3。在本节中,会分别对这两个工具类作出说明。
其中模版引擎类需要引入org.freemarker包来调用FreeMarker API方法,其中最主要的方法是createDynamicContentAsFile,它需要传入一个模版文件、一个存储着键值对的map对象和最终生成的shell脚本文件路径,通过map传入的对象信息映射到模版中,生成最终的WGS测序流程的分析脚本。下面是具体的模版引擎类方法:
/**
* 这个类使用FreeMarker(http://freemarker.apache.org) FreeMarker是一个模板引擎,这是一个根据模板生成文本输出的通用工具
* (从shell脚本到自动生成的源代码都是文本输出)
*/
public class TemplateEngine {
// 日志记录
private static Logger theLogger = Logger.getLogger(TemplateEngine.class.getName());
// 通常在整个应用生命周期中只执行一次
private static Configuration TEMPLATE_CONFIGURATION = null;
private static AtomicBoolean initialized = new AtomicBoolean(false);
// freemarker templates 存放目录
protected static String TEMPLATE_DIRECTORY = "./wgs-templates";
public static void init() throws Exception {
if (initialized.get()) {
return;
}
initConfiguration();
initialized.compareAndSet(false, true);
}
static {
if (!initialized.get()) {
try {
init();
} catch (Exception e) {
theLogger.error("在初始化阶段,模版引擎初始化错误...", e);
}
}
}
// 初始化配置
private static void initConfiguration() throws Exception {
TEMPLATE_CONFIGURATION = new Configuration();
TEMPLATE_CONFIGURATION.setDirectoryForTemplateLoading(new File(TEMPLATE_DIRECTORY));
TEMPLATE_CONFIGURATION.setObjectWrapper(new DefaultObjectWrapper());
TEMPLATE_CONFIGURATION.setWhitespaceStripping(true);
TEMPLATE_CONFIGURATION.setClassicCompatible(true);
}
/**
* 通过模板和keyValuePairs动态创建shell脚本
*
* @param templateFileName 一个模板文件名,如:script.sh.template,其模板目录已在configuration中指定
* @param keyValuePairs 存储数据模型的<K,V>Map
* @param outputScriptFilePath 生成的脚本文件路径
* @return 一个可执行的Shell脚本文件
*/
public static File createDynamicContentAsFile(String templateFileName,
Map<String, String> keyValuePairs, String outputScriptFilePath) {
if ((templateFileName == null) || (templateFileName.length() == 0)) {
return null;
}
Writer writer = null;
File outputFile = null;
try {
Template template = TEMPLATE_CONFIGURATION.getTemplate(templateFileName);
// 合并数据模型和模板,生成shell脚本
outputFile = new File(outputScriptFilePath);
writer = new BufferedWriter(new FileWriter(outputFile));
template.process(keyValuePairs, writer);
writer.flush();
} catch (IOException e) {
theLogger.error("创建文件失败...", e);
} catch (TemplateException e) {
theLogger.error("freeMarker动态创建shell脚本失败...", e);
} finally {
if (writer != null) {
try {
writer.close();
} catch (IOException e) {
theLogger.error("创建shell脚本,写入文件时出现IO异常...");
}
}
}
return outputFile;
}
}3.4.2 基因测序分析流程模版设计
用模版化的思想来构建全基因组分析流程,对要分析的样本对象用参数来代替,将整个分析流程分为Mapper阶段和Reduce阶段两个模版,在Mapper阶段模版主要完成基因测序的样本下载到本地机器、以及测序阶段中的比对、排序、标记重复、创建比对索引文件和对单个样本的变异检测生成中间阶段的变异检测gVCF文件,最后,将每个脚本生成的gVCF文件上传到上传到HDFS上,供Reduce阶段的分析流程使用。下面是Mapper阶段的分析模版:
#!/bin/bash
# 脚本所需的环境变量定义
export BWA=~/biosoft/bwa/0.7.12
export SAMTOOLS=~/biosoft/samtools/1.0/bin
export GATK=~/biosoft/gatk/3.6/GenomeAnalysisTK.jar
export PICARD=~/biosoft/picard/2.18.2/picard.jar
export HADOOP=~/hadoop/bin
# 定义参考序列
export REF=~/wgs/input/fasta/E.coli_K12_MG1655.fa
# 定义单个节点可利用的线程数
THREAD=4
# 定义单个节点可利用的内存资源
MEMORY=4G
# 定义RG INFO
INFO_RG='@RG\tID:${sample_name}\tPL:illumina\tSM:E.coli_K12_${sample_name}'
echo "############" `date "+%Y-%m-%d %H:%M:%S"` "############"
echo "--- 开始处理样本 ${sample_name} ---"
# 定义模板变量
INPUT_FILE_R1=${sample_name}_1.fastq.gz
INPUT_FILE_R2=${sample_name}_2.fastq.gz
cd /tmp
# 从HDFS上下载测序样本Block块
echo "###COMMAND LINE###:" >&2
echo "$HADOOP/hadoop fs -get /wgsv2/input/fastq/$INPUT_FILE_R1" >&2
time $HADOOP/hadoop fs -get /wgsv2/input/fastq/$INPUT_FILE_R1 && echo "" >&2
echo "###COMMAND LINE###:" >&2
echo "$HADOOP/hadoop fs -get /wgsv2/input/fastq/$INPUT_FILE_R2" >&2
time $HADOOP/hadoop fs -get /wgsv2/input/fastq/$INPUT_FILE_R2
echo "*** 从HDFS上下载样本数据块" && echo "" >&2
# ×××××× 基因测序 ×××××××
# 步骤一 比对
echo "###COMMAND LINE###:" >&2
echo "$BWA/bwa mem -t $THREAD -R $INFO_RG $REF $INPUT_FILE_R1 $INPUT_FILE_R2 | $SAMTOOLS/samtools view -Sb - > ${sample_name}.bam" >&2
time $BWA/bwa mem -t $THREAD -R $INFO_RG $REF $INPUT_FILE_R1 $INPUT_FILE_R2 | $SAMTOOLS/samtools view -Sb - > ${sample_name}.bam && echo "*** 基因比对操作完成" && echo "" >&2
# 步骤二 排序
echo "###COMMAND LINE###:" >&2
echo "$SAMTOOLS/samtools sort -@ $THREAD -m $MEMORY -O bam -o ${sample_name}.sorted.bam ${sample_name}.bam -T PREFIX.bam" >&2
time $SAMTOOLS/samtools sort -@ $THREAD -m $MEMORY -O bam -o ${sample_name}.sorted.bam ${sample_name}.bam -T PREFIX.bam && echo "*** 基因数据排序完成" && echo "" >&2
#rm -f ${sample_name}.bam
# 步骤三 标记重复
echo "###COMMAND LINE###:" >&2
echo "java -jar $PICARD MarkDuplicates I=${sample_name}.sorted.bam O=${sample_name}.sorted.markdup.bam M=${sample_name}.sorted.markdup_metrics.txt" >&2
time java -jar $PICARD MarkDuplicates I=${sample_name}.sorted.bam O=${sample_name}.sorted.markdup.bam M=${sample_name}.sorted.markdup_metrics.txt 1>&2 && echo "*** 对BAM文件进行重复标记" && echo "" >&2
# 步骤四 创建比对索引文件
echo "###COMMAND LINE###:" >&2
echo "time $SAMTOOLS/samtools index ${sample_name}.sorted.markdup.bam" >&2
time $SAMTOOLS/samtools index ${sample_name}.sorted.markdup.bam && echo "*** 对BAM文件创建比对索引" && echo "" >&2
# 步骤五 变异检测 | 生成各个样本的中间变异检测文件gvcf
echo "###COMMAND LINE###:" >&2
echo "java -jar $GATK -T HaplotypeCaller -R $REF --emitRefConfidence GVCF -I ${sample_name}.sorted.markdup.bam -o ${sample_name}.g.vcf" >&2
time java -jar $GATK -T HaplotypeCaller -R $REF --emitRefConfidence GVCF -I ${sample_name}.sorted.markdup.bam -o ${sample_name}.g.vcf 1>&2 && echo "*** 生成样本${sample_name}的中间变异检测文件gvcf" && echo "" >&2
# 将各个样本分区的变异检测gvcf文件上传到HDFS上
echo "###COMMAND LINE###:" >&2
echo "$HADOOP/hadoop fs -put -f ${sample_name}.g.vcf /wgsv2/output/gvcf" >&2
time $HADOOP/hadoop fs -put -f ${sample_name}.g.vcf /wgsv2/output/gvcf && echo "*** 变异检测结果gvcf文件上传到HDFS上" && echo "">&2
rm -f rm -f $INPUT_FILE_R1 $INPUT_FILE_R2 ${sample_name}*
echo "--- 结束处理样本 ${sample_name} ---"
echo "############" `date "+%Y-%m-%d %H:%M:%S"` "############" && echo ""对于Reduce阶段的分析模版,主要是对各个Mapper阶段产生的gVCF变异文件的合并,得到最终总的VCF文件,并将其创建tabix索引后上传至HDFS上存储。下面是具体的Reduce阶段分析模版:
#!/bin/bash
# Author: elon
# Description: 合并所有样本的GVCF文件的模板
# Time: 2018-4-5
# 该脚本需要完成的是对各个样本分区的VCGF文件做一个merge操作
export SAMTOOLS=~/biosoft/samtools/1.0/bin
export GATK=~/biosoft/gatk/3.6/GenomeAnalysisTK.jar
export HADOOP=~/hadoop/bin
# 定义参考序列
export REF=~/wgs/input/fasta/E.coli_K12_MG1655.fa
echo "############" `date "+%Y-%m-%d %H:%M:%S"` "############"
echo "--- 开始合并处理样本 ${sample_name1} ${sample_name2} ${sample_name3} ${sample_name4} ${sample_name4} ${sample_name5} ${sample_name6} ${sample_name7} ${sample_name8} ${sample_name9} ---"
# 定义模板变量
INPUT_FILE1=${sample_name1}.g.vcf
INPUT_FILE2=${sample_name2}.g.vcf
INPUT_FILE3=${sample_name3}.g.vcf
INPUT_FILE4=${sample_name4}.g.vcf
INPUT_FILE5=${sample_name5}.g.vcf
INPUT_FILE6=${sample_name6}.g.vcf
INPUT_FILE7=${sample_name7}.g.vcf
INPUT_FILE8=${sample_name8}.g.vcf
INPUT_FILE9=${sample_name9}.g.vcf
cd /tmp
# 从HDFS上download所有的GVCF文件
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE1
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE2
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE3
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE4
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE5
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE6
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE7
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE8
$HADOOP/hadoop fs -get /wgsv2/output/gvcf/$INPUT_FILE9
echo "*** 从HDFS上获取所有相关的gVCF文件"
# 合并所有的gVCF文件为VCF文件 -- E_coli_K12.vcf
echo "###COMMAND LINE###:" >&2
echo "java -jar $GATK -T GenotypeGVCFs -nt 4 -R $REF --variant $INPUT_FILE1 --variant $INPUT_FILE2 \
--variant $INPUT_FILE3 --variant $INPUT_FILE4 --variant $INPUT_FILE5 --variant $INPUT_FILE6 \
--variant $INPUT_FILE7 --variant $INPUT_FILE8 --variant $INPUT_FILE9 \
-o E_coli_K12.vcf" >&2
time java -jar $GATK -T GenotypeGVCFs -nt 4 -R $REF --variant $INPUT_FILE1 --variant $INPUT_FILE2 \
--variant $INPUT_FILE3 --variant $INPUT_FILE4 --variant $INPUT_FILE5 --variant $INPUT_FILE6 \
--variant $INPUT_FILE7 --variant $INPUT_FILE8 --variant $INPUT_FILE9 \
-o E_coli_K12.vcf 1>&2 && echo "*** 合并所有的gVCF文件为VCF文件" && echo "">&2
## 1.将vcf文件压缩
echo "###COMMAND LINE###:" >&2
echo "bgzip -f E_coli_K12.vcf" >&2
time bgzip -f E_coli_K12.vcf && echo "*** 将vcf文件进行压缩" && echo "">&2
## 2.构建tabix索引
echo "###COMMAND LINE###:" >&2
echo "$SAMTOOLS/tabix -p vcf E_coli_K12.vcf.gz" >&2
time $SAMTOOLS/tabix -p vcf E_coli_K12.vcf.gz \
&& echo "*** 给压缩文件构建tabix索引" && echo "">&2
## 3. 上传最终的VCF文件以及索引文件到HDFS上
echo "###COMMAND LINE###:" >&2
echo "$HADOOP/hadoop fs -f -put E_coli_K12.vcf.gz* /wgsv2/output/vcf" >&2
time $HADOOP/hadoop fs -f -put E_coli_K12.vcf.gz* /wgsv2/output/vcf \
&& echo "*** 变异检测结果vcf文件上传到HDFS上" && echo "">&2
#rm -f E_coli_K12.vcf* *g.vcf*
echo "--- 结束合并处理样本 ${sample_name1} ${sample_name2} ${sample_name3} ${sample_name4} ${sample_name4} ${sample_name5} ${sample_name6} ${sample_name7} ${sample_name8} ${sample_name9} ---"
echo "############" `date "+%Y-%m-%d %H:%M:%S"` "############" && echo ""3.4.3 基因测序与模板引擎的结合
在我的项目框架中,对于不同的样本文件,都是采用同一套基因测序分析流程,而测序流程都是通过Linux命令行界面,因此需要通过一定的转换,将这一套分析流程提炼成一个Shell模版,对于不同的样本文件生成和它相应的分析脚本文件,这样就能达到不同的样本使用这同一套分析流程的目的,最后在不同的map任务中分别执行每个样本的分析脚本。测序流程与分析脚本的模版化如图3-2所示。

图3-2 测序流程与分析脚本的模版化
3.5 本章小结
在本章中,主要对基于Hadoop基因测序数据处理的关键技术作了一个剖析和研究,解决了测序流程如何与MapReduce程序结合的问题,以及对于测序流程中各个步骤产生的数据结果如何存储,是存储在本地还是分布式存储系统中的问题,以及对分析流程的完整行作了简要分析,提出了通过设置驱动程序的参数达到对每一行数据分为一个Map程序处理的设计方案,以及通过引入第三方FreeMarker框架来保证分析流程的统一性和完整性问题,最后构建FreeMarker引擎以及对测序流程进行模板化的设计等。
在下一章节中,对基于Hadoop大数据的基因测序平台的流程搭建进行设计与实现。
- Samy Ghoneimy, Samir Abou El-Seoud. A MapReduce Framework for DNA Sequencing Data Processing[D]. British University 2017. ↩
- Apache. Apache FreeMarker™[EB/OL]. https://freemarker.apache.org/, 2018. ↩
- Mahmoud Parsian. 数据算法(Hadoop/Spark大数据处理技巧)[M]. 苏金国,杨健康等译. 清华大学出版社 第四版 2016. ↩














 4497
4497











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









