



网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
[root@iZwz9bhan5nqzh979qokrkZ ~]# cat a.txt
root❌0:0:root:/root:/bin/bash
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
root❌0:0:root:/root:/bin/bash
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
实例一:去掉重复的行
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -u a.txt
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
root❌0:0:root:/root:/bin/bash
注:sort的-u选项它的作用很简单,就是在输出行中去除重复行。
实例二:sort的-n、-r、-k、-t选项的使用
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -t “:” -k 3 a.txt
root❌0:0:root:/root:/bin/bash
root❌0:0:root:/root:/bin/bash
bin❌1:1:bin:/bin:/sbin/nologin
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
注:以":"为分割符的第三列排序,默认从小往大
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -t “:” -k 3 -r a.txt
daemon❌2:2:daemon:/sbin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
bin❌1:1:bin:/bin:/sbin/nologin
bin❌1:1:bin:/bin:/sbin/nologin
root❌0:0:root:/root:/bin/bash
root❌0:0:root:/root:/bin/bash
注:以":"为分割符的第三列排序,从大往小。-r:取反
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -t “:” -k 3 -n a.txt
root❌0:0:root:/root:/bin/bash
root❌0:0:root:/root:/bin/bash
bin❌1:1:bin:/bin:/sbin/nologin
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
注:-n:按数值大小排序,默认从小往大。
[root@iZwz9bhan5nqzh979qokrkZ ~]# cat a.txt | sort -t “:” -k 1.2 -r
root❌0:0:root:/root:/bin/bash
root❌0:0:root:/root:/bin/bash
bin❌1:1:bin:/bin:/sbin/nologin
bin❌1:1:bin:/bin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
daemon❌2:2:daemon:/sbin:/sbin/nologin
注:以":"为分隔符,以第一列的第二个字符排序,默认从前往后,从小到大。
补充:
cat a.txt | sort -t ‘’:‘’ -k 2 以分割符的第2列顺序排序
cat a.txt | sort -t “:” -k 1.2 以分割符的第一列的第2个字符排序
cat a.txt | sort -t “:” -k 1.2 -r 以分割符的第一列的第2个字符倒序排序 replace
cat a.txt | sort -n 按数值排序
cat b.txt | sort -t ": " -k 1.2 -n 以分割符的第1列的第2个字符的数值大小排序
实例三:把排序结果输出到原文件中,用重定向可就不行了
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -nr b.txt
86
79
46
33
20
13
12
11
09
[root@iZwz9bhan5nqzh979qokrkZ ~]# sort -nr b.txt -o b.txt
[root@iZwz9bhan5nqzh979qokrkZ ~]# cat b.txt
86
79
46
33
20
13
12
11
09
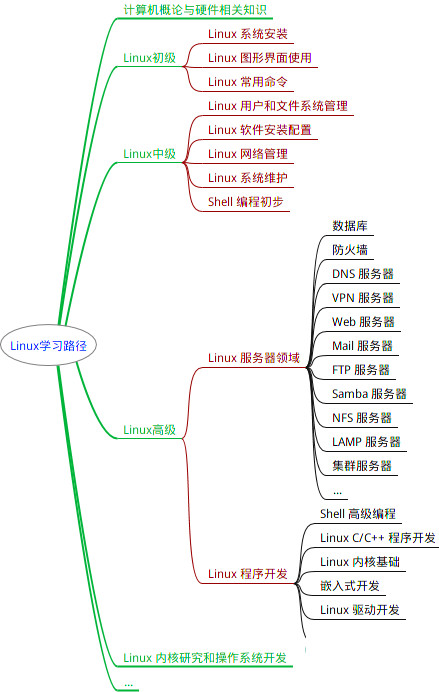
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!

















 5万+
5万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









