









背景介绍
在生物信息学中,FASTA格式(又称为Pearson格式),是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来编码,且允许在序列前添加序列名及注释。(来自百度百科)
其实fasta格式就是简单的文本格式,用C语言中可以用其提供的标准库函数对其进行操作,本文只简单介绍代码中所用到的函数;
文件的打开(fopen函数),函数原型:FILE * fopen(const char * path,const char * mode);打开文件实际是建立文件的联系,使文件指针指向该文件
文件的关闭(fclose函数),函数原型:int fclose( FILE *fp );关闭一个流,注意:使用fclose()函数就可以把缓冲区内最后剩余的数据输出到磁盘文件中,并释放文件指针和有关的缓冲区。
文件读(fread函数)函数原型:size_t fread(void* buff,size_t size,size_t count,FILE* stream);作用:从文件中读入数据到指定的地址中
文件重定位(rewind函数),函数原型:void rewind(FILE *stream); 作用:将文件内部的位置重新指向一个流数据流文件的开头文件定位(fseek函数),函数原型:int fseek(FILE *stream, long offset, int fromwhere); 作用:定位流数据流文件上的文件内部位置指针
程序功能介绍
本程序完成fasta文件的读取,并读取其中核酸序列以及一小段描述符,并将其地址存取于指针变量中,方便对其中每个字符进行操作,
下面贴上代码和注释。
#include <stdlib.h>
#include <stdio.h>
//#define NUM 10*1024
#define ERROR -1
char *buff;
char *start_point;
int size;
char *get_gi(char *p,int n){
static char *m,*t;
m=(char *)malloc(n);
t=(char *)malloc(n);
if(m==NULL||n==NULL)
return NULL;
t=m;
while(n--){
*m++=*p++;
}
*(m)='\0';
return t;
}
char *get_seq(char *p,int n){
static char *m,*t;
m=(char *)malloc(n-1);
t=(char *)malloc(n-1);
if(m==NULL||n==NULL)
return NULL;
t=m;
p=p+n;
while((n<size)&&(*p)){
*m++=*p++;
n++;
}
*(m-1)='\0';
return t;
}
int main(int argc, char* argv[])
{
FILE *file;
size=0;
file = fopen(argv[1],"rt");
/*argv[1]为命令行传入的地址,"rt"表示以只读的方式打开一个文本文件*/
if(file==NULL)
{printf("ERROR:cannot retrieve this file.\n");
return ERROR;}
fseek(file,0L,SEEK_END);
size=ftell(file);
/*通过定位到文件末尾,读出文件大小size,或者也可通过下面注释掉的for循环读取文件大小size*/
rewind(file);
//char a;
//a='0';
//int ii;
//ii=0;
/*for(;a!=EOF;){
a = getc(file);
printf("a is %c%d\n",a,ii);
ii++;
}*/
//rewind(file);
//printf("ii number is %d\n",ii);
printf("The file size is %d\n",size);
buff = (char *)malloc(size-1);
start_point = (char *)malloc(size-1);
if(buff==NULL||start_point==NULL)
return ERROR;
fread(buff,size-1,1,file);
/*将file指向的文本文件内容读入buff缓冲区中*/
start_point=buff;
/*start_point用于存储buff指向的首地址,用于free等*/
printf("%s\n",buff);
/*打印出文本文件内容,此处用于调试,printf是个很好的调试方法,此处可检查文本是否读出,以及是否正确等*/
static int i;
//unsigned short *aa;
//printf("%c\n",*buff);
static int pos;
static int seq_pos;
for(;*buff;buff++){
//printf("%p\n",buff);
i++;
if((*buff=='|')&&(*(buff+1)==' ')){
pos=i;
//buff--;
printf("The value of pos is %d\n",pos);
}
if((*buff=='A'||*buff=='T'||*buff=='C'||*buff=='G')&&(*(buff+1)=='A'||*(buff+1)=='T'||*(buff+1)=='C'||*(buff+1)=='G')\
&&(*(buff+2)=='A'||*(buff+2)=='T'||*(buff+2)=='C'||*(buff+2)=='G')){
seq_pos=i-1;
printf("The value of seq_pos is %d\n",seq_pos);
break;
}
}
/*for循环中记录了标识符的结束位置和核酸序列的起始位置,这里的标识符是指的第一个空格前面的字符*/
//printf("%d\n",i);
char *mm=get_gi(start_point,pos);
/*mm指向标识符的首地址,被调函数get_gi其实是用指针变量实现的字符串的拷贝,末了需要加上结束符'\0'*/
char *seq=get_seq(start_point,seq_pos);
/*seq指向核酸序列的首地址,被调函数get_seq也是指针变量实现的字符串的拷贝*/
printf("DESCRIPTOR=\n%s\n",mm);
printf("SEQ=\n%s\n",seq);
buff=start_point;
free(buff);
//free(mm);
//free(seq);
fclose(file);
//getchar();
return 0;
}编译执行
在Linux下编译通常通过makefile进行,它的一个好处就是自动化编译,文章此处不再赘述,网上关于makefile帖子非常多,我贴上这次用到的一个很简单的makefile ,源文件为seq.c,makefile注意和源文件在一个目录。
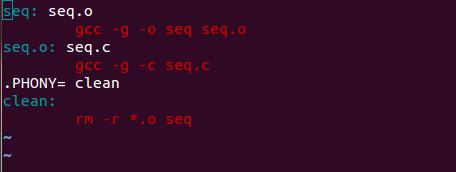
终端切换到当前目录下make,编译成功后会形成一个二进制可执行文件和中间目标文件seq.o

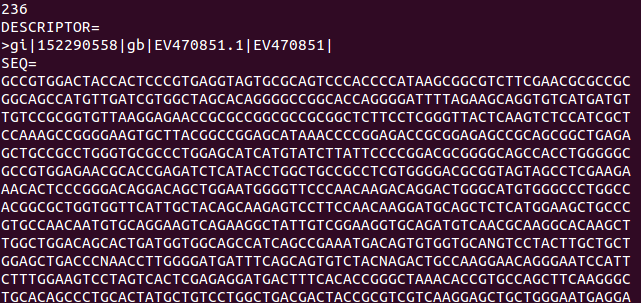
执行seq可执行文件,并传入要读取的文件

成功读出序列和标识符
结束语
C语言其实远没有想的简单,指针真的是C语言的灵魂所在,想要真正用好指针不容易。在python中读取这个序列很简单,就是调用模块的一个函数,但C语言总能让我们思考的更多,它不像python,java,标准库函数中可供我们调用的很有限,几乎每一步都要自己实现,很好地体现了面向过程的思想。














 1969
1969











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









