







序列比对概念及相应工具的对比
0x01 概念和意义
在生物信息学研究中,最基本的部分是序列比对,而最基本的操作是比对,主要可分为双系列比对和多序列比对,辨别序列之间的差异,同时发现生物序列中的机构和功能信息,进而发现它们的相似性和同源性,比较多个生物序列相似性是由序列比对来完成的。总的来说,序列比对的意义是对序列的相似性在核酸、氨基酸的层次上进行分析,从而推测比对中的各个序列间结构功能以及进化上的联系,它也通过对各种不同类型的生物序列进行比对,以寻找与确定比对序列的稳定区域与变化规则,发现它们的功能特征和区别。通过序列比对,也可以检测新序列与数据库中已知序列的相似性关系(结构和功能),从而为确定新序列的结构和功能信息提供事实根据,由上可说明序列比对是基因识别、分子进化、生命起源等研究的基础,而对序列比对的研究于基因结构和功能的研究具有较大的实际意义。
研究序列相似性的目的之一是通过相似的序列得到相似的结构或相似的功能,另一个目的是通过序列的相似性,判别序列之间的同源性,推测序列之间的进化关系。
相应地在恶意代码api序列检测上也可以适用。
序列比对又称序列重排或对准,根据进行比对的生物序列数目序列比对可分为双序列比对和多序列比对。
双序列比对算法点阵图方法和动态规划算法,而多序列比对算法主要 有渐进比对和迭代比对
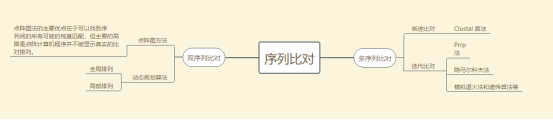
双序列比对主要有:FASTA 和 BALST算法
FASTA算法(使用候选区域)
首先,需找待查序列与已知序列长度为 k 的公共子串,命名为热点区域;
第二,延长热点区域,形成更长的部分比对区域;
第三,综合第二步的比对区域,获得一个得分更高的比对;
最后,基于上述的比对片段,寻找另一个备选的比对。
近似寻优,其缺陷是结果的最优比对无法保证。
BLAST 算法(搜索算法和搜索结果的统计学评估)
第一步,寻找查询序列与靶序列之间长度为 k 的匹配片段;
第二步, 筛选相距较远的匹配片段;
第三步, 向两端延长匹配片段,形成更长的比对区域,在延长过程中,若得分超过某个阈值,则称这些区域为高得分区域,所得的高得分区域按降序排列后作为算法的输出。
在生物序列分析中,有时需要识别多条序列的公共特征,这就要进行多序列的最佳比对分析。
- clustalW。clustalW 是应用最为广泛的多序列比对算法,它采用渐进式比对算法,实现过程主要分为三个步骤:第一,对所有的序列进行两两比对,并构建一个距离矩阵。第二,从距离矩阵出发生成一个用于比对的指导树。第三,根据指导树给定的序列次序,将每个序列依次加入,逐渐形成有更多序列参与的比对,直到所有序列都加入,最后输出最终的比对结果。
- YAMA。YAMA 是一个用于比对长度较长的 DNA 序列的算法,也采用了渐进式的比对方法,在每一次的比对中,设法使两两组对求和值达到最大化,该算法将比对区域限制在动态规划矩阵的两条边界之间来提高计算效率。在进行比对时,可以预先设定一组匹配模式,YAMA 算法就会从最终得到高分比对中选择出跟先前设定模式相匹配的结果。
- MUSCLE。MUSCLE 也是一个高效的多序列比对工具,首先,基于两序列比对的得到的相似性程度构建距离矩阵,然后在距离矩阵的基础上生成进化指导树;然后,在第一步指导树的基础上形成初步的序列多序列比对,由此可以获得每两条序列包含相同残基的概率,并计算其距离,然后根据此距离再次获得进化指导树,此过程又称精化指导树;最后,在精化指导树的基础上做删除处理,从而找到更优的新比对结果,如此将该过程反复进行,直到达到最优的比对结果。
0x02 序列比对及相似性
本节转载自https://blog.csdn.net/weixin_43202635/article/details/82962032
- 序列表示
考虑使用蛋白质序列

2.序列的相似性
数据库中的序列相似性搜索——相似的序列可以推测出相似的结构,相似的结构可以推测出相似的功能。
用两个指标来描述序列的相似性——一致度和相似度。

引出问题:残基的相似如何量化?
残基两两相似的量化关系由替换记分矩阵所定义。


注:密码子——密码子(codon)是指信使RNA分子中每相邻的三个核苷酸编成一组,在蛋白质合成时,代表某一种氨基酸的规律。信使RNA在细胞中能决定蛋白质分子中的氨基酸种类和排列次序。信使RNA分子中的四种核苷酸(碱基)的序列能决定蛋白质分子中的20种氨基酸的序列。而在信使RNA分子上的三个碱基能决定一个氨基酸。


3.比对两个序列的方法
(1)打点法


(2)序列比对法

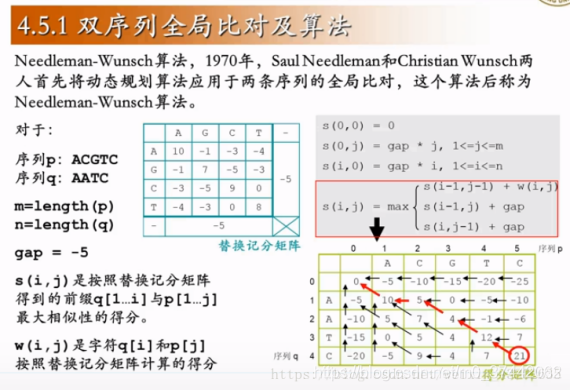
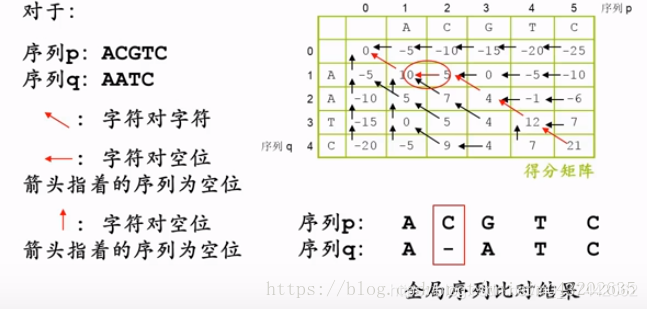
- 双序列序列全局比对算法(用于比较两个长度近似的序列)
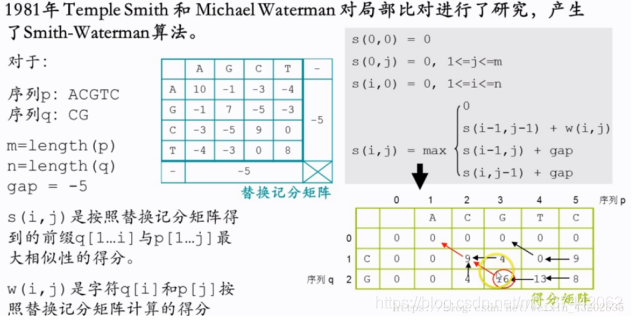
B)双序列局部对比算法(用于比较一长一短两条序列或者是非同源序列)

**
*序列比对算法总结:
 *在线序列比对工具: EMBL、PIR 等
*在线序列比对工具: EMBL、PIR 等

三.多序列比对
多序列比对——对两条以上的生物序列进行全局比对
注:蛋白质家族(英语:Protein family)是一组与进化相关的蛋白质。家族中的蛋白质来自共同的祖先(见同源),通常具有相似的三维结构,功能和显着的序列相似性。
1.多序列比对的主要用途及算法

多序列比对的算法:
目前所有的多序列比对工具都不是完美的,它们都是使用一种近似的算法。
2.多序列比对工具
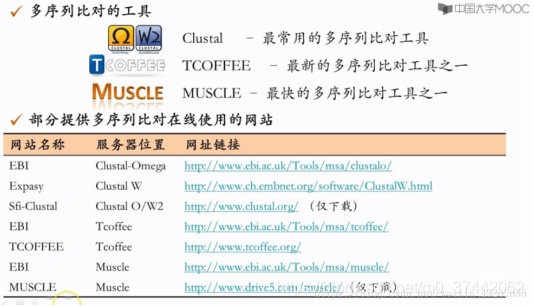
注:1.保守序列在生物学中是指在核酸序列(如RNA及DNA序列)、蛋白质序列、蛋白质结构或多聚糖序列内相似或相同的序列,这种情况可以发生在各物种间(种间同源序列)或由相同生物产生的不同分子(种内同源序列)间。
2. 系统发生树又称演化树或进化树(evolutionary tree),是表明被认为具有共同祖先的各物种间演化关系的树状图。是一种亲缘分支分类方法(cladogram)。在图中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)。
3.寻找保守区域
(1)序列标识图Weblogo

(2)序列基序MEME

尝试找一找基序??
(3)蛋白质指纹PRINTS

家族的指纹图谱
四.补充
1.分子进化理论(未被证实正确)

2.同源
同源序列——来源于共同祖先的相似的序列,分为直系同源、旁系同源、异同源
注意:相似序列不一定是同源序列。
相似度可以量化,同源性不可量化,它只是对性质的一种判定。
3.系统发生树
系统发生树又称演化树或进化树(evolutionary tree),是表明被认为具有共同祖先的各物种间演化关系的树状图。是一种亲缘分支分类方法(cladogram)。在图中,每个节点代表其各分支的最近共同祖先,而节点间的线段长度对应演化距离(如估计的演化时间)。
构建系统发生树的意义:
对于一个位置的基因或蛋白质序列,确定其亲缘关系最近的物种;
预测一个新发现的基因或蛋白质的功能;
有助于预测一个分子功能的走势。
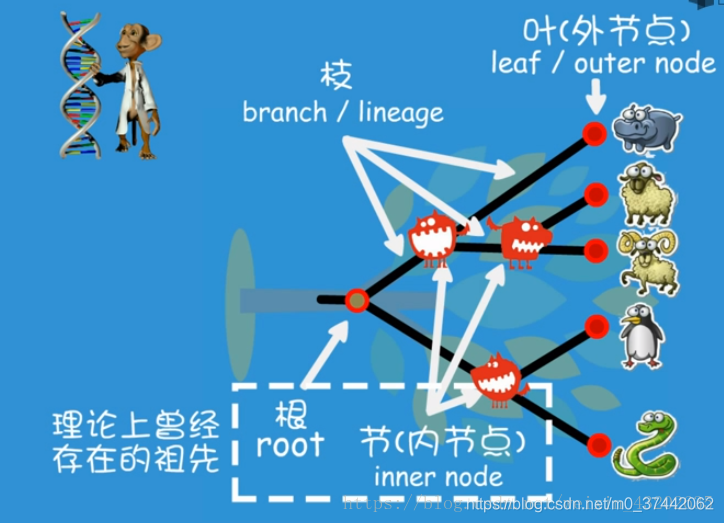
注意:根、内节点都理论上曾经存在过的共同祖先,现在已经没有了
参考资料
慕课课程:https://es.coursera.org/lecture/sheng-wu-xin-xi-xue/cong-quan-ju-bi-dui-dao-ju-bu-bi-dui-2QeSA
序列比对
完成本模块的课程后你将可以: 掌握基于动态规划编程思想的序列比对算法; 区分Needleman-Wunsch全局比对算法和Smith-Waterman局部比对算法; 了解空位罚分背后的原理和计算算法的复杂度将帮助你在你自己的研究中应用现有的生物信息学工具; 你还可以一睹Smith-Waterman算法的发明人Michael Waterman博士的风采。
可以跟着做的实验:
生物信息之多序列比对,进化树分析,保守位点分析
https://blog.csdn.net/u011262253/article/details/78506951
黄佳琪. 生物信息学序列比对算法分析[J]. 生物技术世界, 2015(11):279-279.














 2398
2398











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









