







论文来源:
Prediction of Drug-Likeness Using Deep Autoencoder Neural Networks
摘要
由于各种各样的原因,大多数候选药物最终不能成为上市药物。建立可靠的预测候选化合物是否具有药物相似度对提高药物的发现和开发成功率具有重要意义。
在这篇文章中,做事使用全连接神经网络(FNN)来构建类药性分类模型,并使用深度自编码器来初始化模型参数。
收集了药物(以ZINC World Drug为代表)、生物活性分子(以MDDR和WDI为代表)、普通分子(以ZINC All Purchasable 和ACD为代表)的数据集。
化合物用MOLD2二维结构描述符编码。类药/非类药模型在WDI/ACD数据库中的分类准确率为91.04%,在MDDR/ZINC数据库中的分类准确率为91.20%。模型的性能优于以前报告的模型。
此外,我们还开发了药物/非药物类模型(锌世界药物vs.锌全可购),用于区分药物和常见化合物,分类准确率为96.99%。我们的工作表明,通过使用高纬度的分子描述符,我们可以应用深度学习技术建立最先进的药物相似度预测模型。
一、简介
近几十年来,高通量筛选(high-throughout screening, HTS)、基于片段的药物发现(fragment- based drug discovery, FBDD)、单细胞分析等多种新颖有效的技术得到了发展,并在药物发现领域取得了显著进展。然而,值得注意的是,FDA批准的新化学实体(NCEs)的数量并没有像预期的那样快速增长。据统计,在临床前检测中发现候选化合物的成功率约为40%,而进入市场的候选化合物仅为10% (Lipper, 1999)。
约有40%的候选化合物没有上市是因为它们的生物制药性能差,也通常被称为药物相似性,包括化学稳定性差、溶解性差、渗透性差和代谢差(Venkatesh and Lipper, 2000)。药物相似性是由现有药物和候选药物的结构和性质衍生而来的,在药物发现的早期阶段被广泛用于筛选不良化合物。
最初的药物如Lipinsky提出的规则的概念,称为rule-of-five包含四个简单的物理化学参数定义(MWT≤500,log P≤5,H-bond donors≤5 ,H-bond acceptors≤10)(Lipinski,2004)。使用这些定义可以预测一种化合物是否可以成为口服药物的候选。
Hopkins等人在2012年提出了药物相似度定量估计(QED)测度,该测度是一种加权可取度函数,它是基于一组771种口服小分子药物的8种选定分子性质的统计分布,应用于分子靶向给药能力评估(Bickerton et al., 2012)。由于药物与非药物的分子性质定义不明确,且描述符较少,预测效果不理想,后来的工作试图结合较为全面的描述符和大量的化合物数据,从定量的角度建立高准确度的药物相似度预测模型。
前人研究成果
Wagener等人提出了一种药物相似度预测模型,涉及到与不同原子类型数目相关的分子描述符和用于区分潜在药物和非药物的决策树。模型使用来自ACD和WDI的10,000个化合物进行训练,其在177,747个化合物的独立验证数据集上的预测ACC为82.6% (Wagener and van Geerestein, 2000)。在2003年,Byvatov和他的同事使用不同的描述符集和描述符组合来表征化合物,并应用SVM和人工神经网络(ANN)系统来解决药物/非药物的分类问题。两种方法的预测准确率均达到80%,结果表明支持向量机的鲁棒性更强(Byvatov et al., 2003)。Muller后来报道的一个模型也是基于支持向量机的,模型选择过程非常仔细,以提高Byvatov et al.(2003)的预测结果(Muller et al., 2005)。2008年,Li等人实现了ECFP_4 (Extended Connectivity fingerprint,扩展连通性指纹图谱)对分子进行表征,并使用概率支持向量机模型对类药物分子和非类药物分子进行分类。与之前在相同数据集上的工作相比,该模型显著提高了预测ACC,令人惊讶的是,在使用341,601个化合物的更大数据集时,分类器将ACC提高到了92.73% (Li et al., 2007)。Schneider等人利用决策树,基于SMARTS字符串、分子量、XlogP和分子折射率作为化合物的描述空间,逐步对类药物化合物进行硅酸盐筛选(Schneider et al., 2008)。2012年,Tian等人实现了21种理化性质和LCFP_6指纹编码分子,利用朴素贝叶斯分类(naive Bayesian classification, NBC)和递归划分(recursive partitioning, RP)构建类药物/非类药物分类器,实现了90.9%的ACC (Tian et al., 2012)。这些研究表明,机器学习技术结合大数据集在药物相似度预测问题上具有很大的潜力。
深度学习是基于人工神经网络(ANN)的机器学习新浪潮。自2006年以来,DL在计算机视觉等诸多领域表现优异(Hinton et al., 2006;Coates等,2011;Krizhevsky等,2012;He et al., 2016)自然语言处理(Dahl et al., 2012;Socher等,2012;Graves等,2013;Mikolov等,2013;生物信息学和化学信息学(Di Lena et al., 2012;Lyons et al., 2014;Heffernan等,2015;陈等,2016;曾等,2016)。与传统的机器学习方法相比,多层DL可以自动将原始数据转化为合适的内部特征表示,有利于检测或分类任务(LeCun et al., 2015)。在本研究中,我们使用深度自编码器神经网络构建了强大的药物相似度预测模型,并从MDDR、WDI 和ZINC。通过Mold2 (Hong et al., 2008)和Padel (Yap, 2011)计算化合物的分子描述子。类药/非类药模型在WDI / ACD数据库中的分类准确率为91.04%,在MDDR /ZINC数据库中的分类准确率为91.20%。模型的性能优于以前报告的模型。此外,我们开发了药物/非药物样模型(ZINC World drug vs. MDDR),用于区分药物和常见化合物,ACC分类为96.99%。我们的工作表明,通过使用高纬度的分子描述符,我们可以应用DL技术建立最先进的药物相似度预测模型。
二、数据集
2.1基准数据集
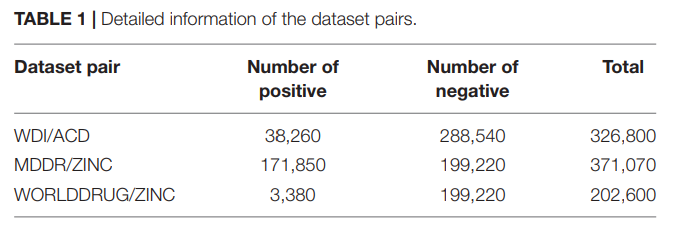
本研究将整个化学空间分为药物、类药物和非类药物。上市药物分子以 ZINC WORLD drug (Sterling and Irwin, 2015) (version 2015, 2500 molecules)数据集为代表。类药物分子 用MDDR (MACCS-II药物数据报告[MDDR], 2004) (200k分子)数据集和WDI (Li et al., 2007)(2002版,40k分子)数据集表示。非药物样分子用ACD (Li et al., 2007) (version 2002, 300k molecules)和ZINC ALL PURCHASABLE(Irwin et al., 2012) (version 2012)数据集表示;后者被随机取样,减小到200k。类药物数据集包含已上市和类药物分子,而非类药物数据集包含另外两个数据集。所有数据集都包含SDF格式的二维分子结构信息。本研究使用的数据集对的详细信息见表1。
2.2数据预处理
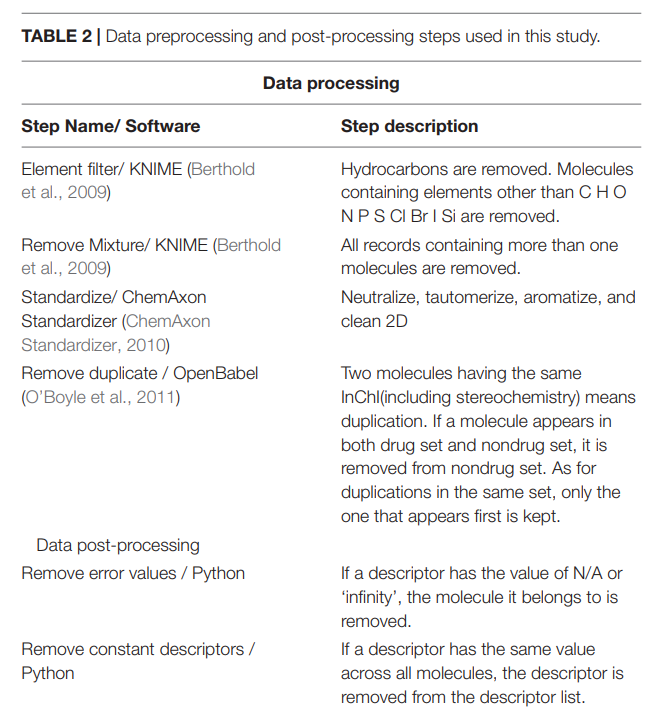
数据清洗是化学信息学计算中的一个关键步骤,Fourches等人(2010)阐述了这一点。我们使用了一个类似于Fourches等人的流程(见表2)来预处理下载的原始数据,从而减少了在描述符计算中出错的可能性。在描述符计算之后,我们还对得到的描述符矩阵进行后处理(见表2)。
2.3描述符计算
我们用二维描述符对分子进行编码。预处理后的分子MOLD2计算,得到一个描述符矩阵∼700每个分子描述符。然后对描述符矩阵进行后处理,如表2所示。我们还尝试了Padel descriptors (Yap, 2011),该描述符在本研究中表现较差,被丢弃。
2.4过采样(Over-Sampling)算法
由于分类任务的特殊性,本研究中我们采集的阳性和阴性样本并不均衡。利用不平衡数据建立的预测模型可能存在偏差和不准确。因此,我们采用两种方法来平衡我们的数据集,使正样本和负样本的比例近似相等。 第一种方法是复制minority类,使其比例为1:1,第二种方法是使用SMOTE (Chawla et al., 2002;Han et al., 2005;(Nguyen et al., 2011),是一种基于随机过采样算法的改进方案。这里我们使用从【1】下载的imbalanced-learn包来应用SMOTE。对于每个任务,我们使用这两种过采样方法来平衡数据。对于每一个模型,我们首先将9:1的比例的数据集随机分割为训练集和验证集,然后利用上述两种方法来平衡训练集,使训练过程中正负样本的数量相等。使用训练集对5-CV模型进行训练,使用附加验证集对模型进行评价。
【1】http://contrib.scikit-learn.org/imbalanced-learn/stable/install.html
三、材料和方法
3.1堆叠自动编码器
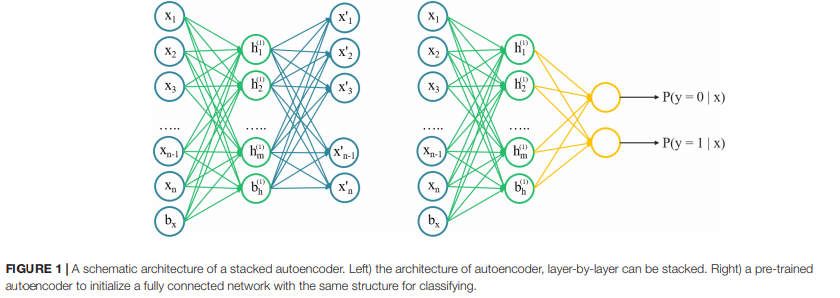
自动编码器是一种无监督的学习算法,它训练神经网络重构其输入,更有能力捕捉输入数据的内在结构,而不仅仅是记忆。直觉上来看,它试图建立一个encoding-decoding过程,模型的输出xˆ大约是类似于输入x。SAE是一个神经网络组成的稀疏autoencoders的多个层,每一层的输出是连续的连接到输入层。SAE的体系结构示意图如图1所示。利用二维化学描述符对声发射模型进行训练,找出描述符之间的内在联系,然后利用声发射模型的参数对分类模型进行初始化。
3.2定义模型
根据化学空间对药物、类药物和非类药物的划分,可分为类药物/非类药物、药物/非类药物两类分类模型。第一个与传统的相似度定义相吻合。第二个模型也具有相当大的实用价值,但是还没有发表任何模型来解决这个问题。
在本研究中,为了解决类药物/非类药物的分类问题,我们提出了两种模型:
模型1:类药物/非类药物的分类问题
MDDRWDI/ZINC(即MDDR和WDI为阳性集,ZINC为阴性集)和WDI/ACD搭建的模型。
模型2:药物/非药物类药物的分类问题
我们提出了WORLD drug/ ZINC(即ZINC WORLD drug为阳性集,ZINC ALL purchasing able为阴性集)模型。
3.3网络训练与超参数优化
在本研究中,我们使用基于Tensorflow (Abadi et al., 2016)的开源软件库Keras (Chollet, 2015)构建SAE模型和分类模型。首先,训练单个隐藏层AE。隐含层节点数K,是需要跨不同网络进行比较和调优的超参数。在训练中,我们使用Truncated-Normal初始化器生成一个截断正态分布层的权重。在所有情况下,我们都使用了贝叶斯优化(Hyperas,一个基于hyperopt的python库【2】)来优化超参数,如隐藏层节点K的个数,L2权值正则化器的值,drop的值,激活函数的类型,优化器的类型,批大小的值。最终的超参数优化设置如表3所示。

考虑到虽然数据集已经平衡,但模型结果可能会过拟合,因此我们对对数似然损失函数的正样本损失和负样本损失的权重进行优化为:
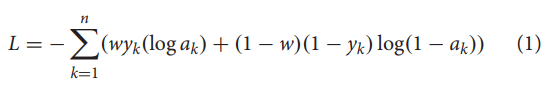
其中yk表示第k个复合标签。yk = 1或0,表示第k个化合物分别为类药物化合物和非类药物化合物。ak = P(yk = 1|xk)为模型计算得到的第k个化合物为类药物化合物的概率。w为正样本损失的权重。在不同的情况下,我们选择最合适的w的范围(0.5∼1.0),以避免过度拟合。然后我们对所有模型进行5- cv的训练,并在测试集上基于分类ACC强制提前停止。最后,每个案例有5个训练模型,取平均值作为这些模型的最终判断。
3.4 模型评价
所有模型均采用5个指标进行评价。分别定义了ACC、SP、灵敏度(SE)、MCC、接收机工作特性曲线下面积(AUC)四个判据,分别为:
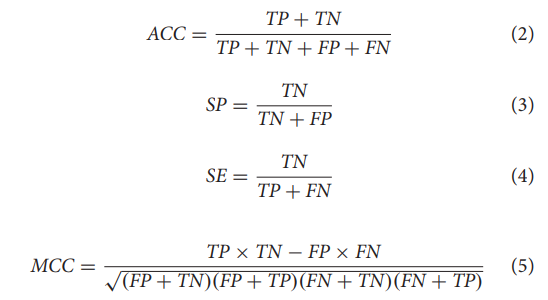
四、结果
4.1比较不同的过采样方法
在我们对5-CV验证测试进行预训练后,我们发现更多的层数和神经元数量并不能提高预测能力。在所有情况下,一个隐藏层足以满足我们的分类目标。通过分析两种不同的过采样方法来平衡数据集,复制少数派类和SMOTE,我们发现后者可以达到更好的预测精度,如表4所示:
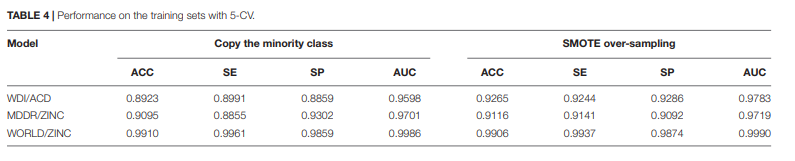
在相同的数据集下,Li等构建的SVM模型的ACC为92.73% (Li等,2007),我们WDI/ACD模型的ACC为92.65%,与Li等的结果基本一致。
我们的MDDRWDI/ZINC模型对类药物/非类药物分子进行了分类,ACC达到了令人满意的91.16%,是目前最先进的类药物预测模型。这些结果表明,自编码器是一种潜在的药物相似度预测机器学习算法。
我们基于World drug/ ZINC数据集建立的药物/非药物类预测模型ACC高达99.06%,说明药物和非药物更容易区分化合物。虽然不排除后一种模型的ACC与原始数据集的严重失衡有关,但我们认为这种药物/非药物类预测模型可能有利于药物开发。
4.2 优化参数和损失函数
我们观察到,使用从原始数据中预先分割出来的独立外部验证集对模型进行评价时,模型的预测ACC往往略低于训练的预测ACC,但敏感性值显著降低,SP值较高(表5),说明模型在训练中存在过拟合现象。
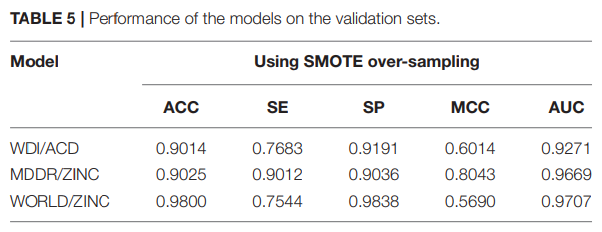
其根本原因可能是原始数据的正样本比过低,我们按照9:1的比例将原始数据集中的正样本和负样本随机分割,构建训练集和验证集。即使使用SMOTE方法来平衡训练集中的正样本和负样本,由SMOTE生成的新阳性样本依赖于原始训练集中的正样本,因此,外部验证集的正样本信息较少。
为了克服负样本的过拟合问题,我们在损失函数中增加了正样本损失的权重,增强了模型对正样本侧的学习能力。我们将权重值(详见公式1)从0.5到1进行测试,每隔20次,记录验证集上ACC、SE、SP值随权重变化的情况,如图2所示。

对于不同的模型,图2曲线中SE和SP的交点对应一个平衡的权值。通过微调(fine-tuning),四种模型对应的权重分别为(0.69、0.55和0.9)。对损失函数使用这些权重后,不同模型下训练集的ACC略有下降,SE有所改善。由于模型增强了对阳性样本的预测,不同模型验证集的SE和SP值接近(见表6,7)。
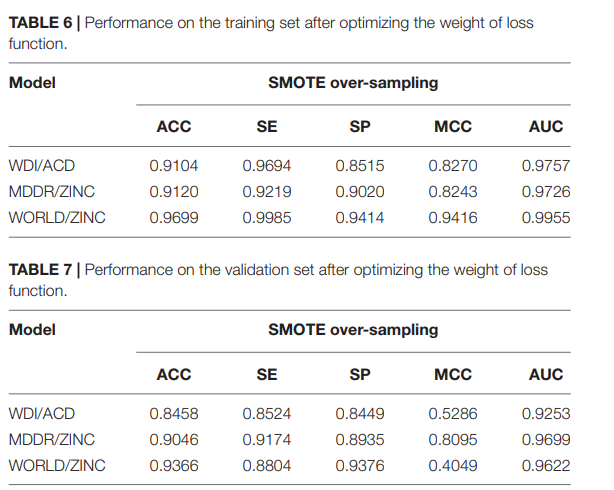
虽然MCC一般被认为是一种平衡测度,但它受到数据集正负样本数的差距和模型计算的混淆矩阵的严重影响。MCC对于平衡训练集是满意的。但在验证集中,数据集变得更加不平衡,MCC变小,这是不可避免的。
五、讨论
在图像识别问题中,当产生AE时,常常堆叠数个AE层以制作SAE。尽管发现SAE比单层AE更强大,但是我们发现SAE在药物相似性问题中是有缺陷的,使得多层SAE的执行比单层AE要差很多。
当对AE的一层进行训练时,期望将输出尽可能地接近其输入,并且该误差可以被定义为输出减去输入的平均值。在这项研究中,当训练模型,我们发现规范化的ACC(z分数)输入是远远高于缩放(−1,1)的输入。对数据进行标准化处理后,AE的误差为0.8,比图像识别中的典型值高了一个数量级。叠加层的AE将进一步放大误差,使SAE-initialized神经网络在分类表现不佳
我们认为AE的这种缺陷源于不同维度的输入数据是如何相互关联的。在图像识别中,每个像素都是一个维度;在药物相似度预测和相关领域,每个描述符都是一个维度。AE的训练目标是学习维度之间的关系,将输入的信息编码成隐含的层维度。因此,如果维度之间的关系本质上更加混乱和不规则,AE很可能会表现得更糟。像素之间的关系是规则的,因为它们被组织成一个二维网格,而相邻的像素具有一些相似性和互补性。描述符之间的关系缺乏这种良好的性质,导致AE输入重建过程失败。尽管AE重建错误很大,但我们的模型仍在分类中表现良好。我们认为这是因为AE预训练的正则化效应。在无监督的训练前,该模型更有能力真正学习数据,而不是简单地记忆数据。
不平衡的数据集是一个常见的问题。虽然有一些方法,如SMOTE,可以生成新的数据来平衡数据集,但是这种生成数据的方法很大程度上依赖于样本的分布。一旦样本分布非常稀疏,新的数据很可能会偏离原始数据被释放的空间。根据已有数据的分布寻找数据映射空间的方法是生成数据以平衡数据集的关键,如当前流行的深度生成模型。开发新的算法来训练不平衡数据集也是一个重要的研究方向。
在这项研究中,DL再次显示了它改进预测模型的能力。尽管取得了成功,但我们认为还有很大的发展空间。一个关键的方面是使当前的DL方法适应特定的问题。这种适应应该基于对当前DL方法的更好理解。也就是说,要知道方法的哪些部分是可以通用的,哪些部分应该根据数据的性质进行修改。例如,在这项研究中,我们认为训练的规范化效应是通用的部分,在输入数据不规则的时候,AE输入重构的部分应该被取消或修改
结论
在这项研究中,我们手工构建了两个更大的数据集,类药物/非类药物和类药物/非类药物。然后用了AE预训练法,我们研发出了药物相似性预测模型。基于WDI和ACD数据库的分类ACC提高到91.04%。我们的模型在MDDRWDI/ZINC数据集上的分类ACC达到了91.20%,是目前最先进的药物相似度预测模型,说明DL模型的预测能力优于传统的机器学习方法。此外,我们开发了药物/非药物样模型(锌世界药物vs.锌全可购),对药物和常见化合物进行了区分,ACC分类为96.99%。我们提出在这项研究中,AE预训练是一种更好的正则化方法。本研究中多层SAE重建的失败表明,由于数据的特殊性,在将DL应用于不同领域时可能需要进行一些修改。我们希望未来机器学习的研究者和化学家能够紧密合作解决这一问题,使DL方法在化学问题中的理解和应用更加深入。














 857
857











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









