










推荐度:⭐⭐⭐⭐⭐
亮点
设计了一种机器学习构架AgentBind,可以识别和解释对于转录因子(TF)结合最重要的序列特征。与以往大多数研究结合基序的系统或程序不同,AgentBind着眼于基序附近的序列背景,并可以研究其在TF结合中的作用。
近日由加利福尼亚大学圣地亚哥分校郑安与Melissa Gymrek等的研究团队在《Nature Machine Intelligence》杂志上发表了一篇名为“Deep neural networks identify sequence context features predictive of transcription factor binding”的研究文章。转录因子(TF)通过识别特定的序列基序来结合DNA,。一个基序可以在人类基因组中发生数千次,但实际上只有这些位点的一部分被结合。这篇文章作者构建了一种名为“AgentBind”机器学习构架,可用于识别对于TF结合最重要的序列特征。研究人员使用这一构架预测在淋巴母细胞细胞系中38个转录因子的基序结合,评估了特定碱基对于序列的重要性,并表征最能预测结合的序列特征。这项研究为人们预测转录因子结合的特征提供新颖的见解,并有可能为将来的深度学习应用提供信息,以解释非编码遗传变异。

转录因子(TFs)的结合是主要的转录调控机制之一,TFs识别并结合特定的DNA序列以控制转录,这一过程形成了指导基因表达的复杂系统。 TF结合的破坏是包括癌症、自身免疫性疾病、心血管疾病等多种疾病发病的根本原因之一。因此,了解控制TF结合的机制可以深入了解疾病过程,为开发新的治疗手段提供思路。研究表明,大多数TF具有独特的结合偏好,只能识别包含特定模式(即核心基序)的DNA序列。但是基序匹配序列和实验确定的结合位点之间通常只有部分重叠。特定基序是否能够与TF结合取决于许多其他因素,包括染色质可及性,核小体定位,与其他TF的协同和竞争结合等等。这些因素中有许多与TF基序周围的序列背景有关。为了研究序列上下文在TF结合中的作用,我们开发了一个名为AgentBind的构架,用于预测是否会结合基序实例,以及寻找对结合状态影响最大的特定核苷酸。
以下是本文第一作者郑安对于该技术的解读:
该模型架构包含三个步骤:预演,微调,解释(pre-training, fine-tuning, interpretation),并应用DanQ作为模型体系结构。首先利用pre-train DanQ模型从多个细胞获取外遗传性注释类型。随后将为每个TF建立一个二进制数据集:提取以基序实例为中心的1kb基因组序列,并根据与ChIP测序确定的结合位点重叠将每个序列标记为结合(阳性)与未结合(阴性)。每个二进制数据集用于微调一个单独的预演模型,使其学习TF绑定的重要功能。最后将使用一种名为Grad-CAM的模型解释方法对每个核苷酸对结合预测的贡献进行评分(图1)。
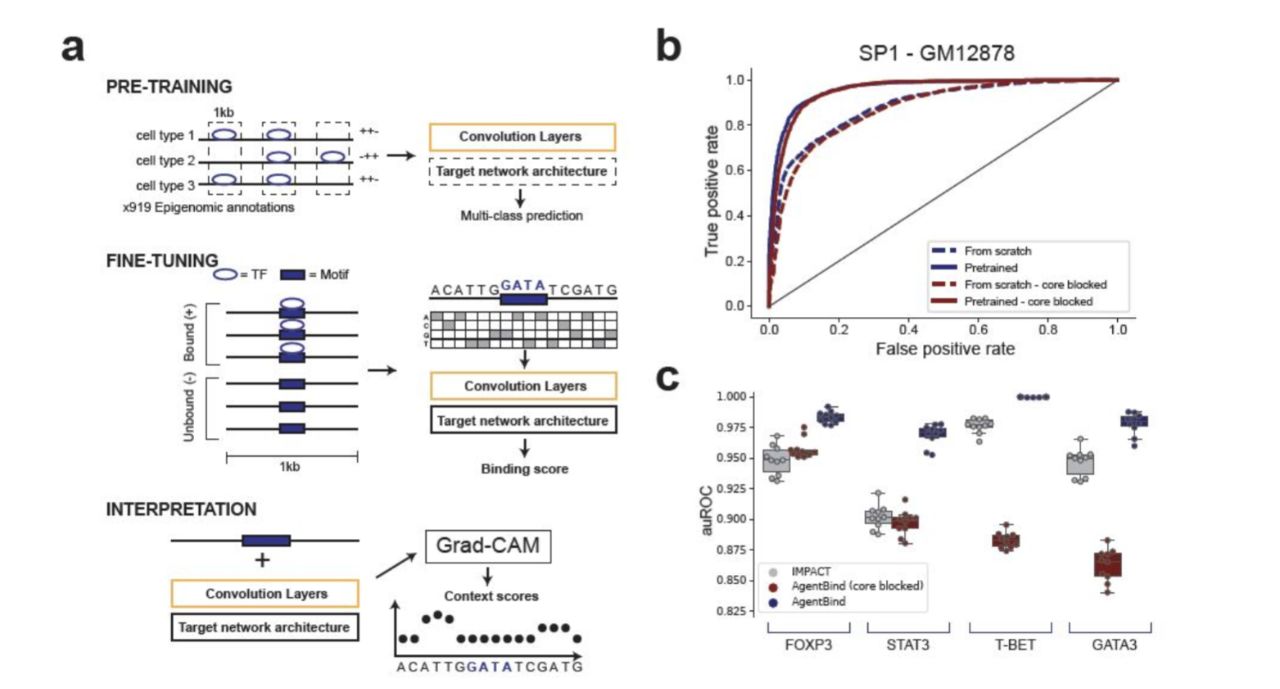
随后研究人员用这一架构重点研究了淋巴母细胞样细胞系GM12878,其中有
活跃的38个TF,具有可从ENCODE项目获得的ChIP-seq数据集,以及可从JASPAR获得的结合位点基序。通过AgentBind,研究人员确定了预测转录因子结合的核苷酸碱基。图2显示了包含某个区域的Grad-CAM分数的示例,y轴显示了每个核苷酸的Grad-CAM分数,结果显示中央SP1基序的序列和对应于NFY基序的两个高分区域。
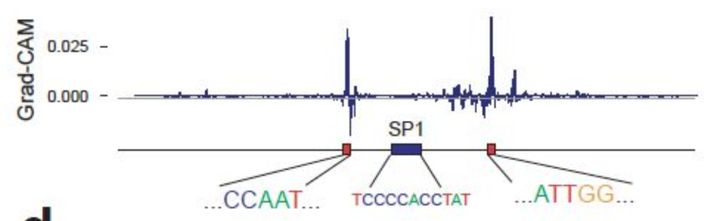
对于阴性样本,研究人员可以选择将它们限制在DNase I超敏感位点之内或之外,从而产生两个不同的模型:DNase-I受控模型和基线模型。图3显示了这两个模型确定的关键上下游序列特征。虽然基线模型可产生更好的分类结果,但DNase-I控制的模型识别出基线模型中忽略的一些不同模式。

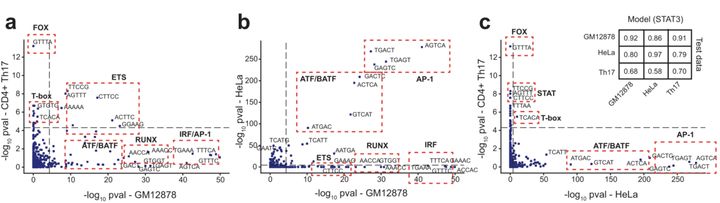
为了研究这一框架捕获细胞类型特异性调控功能的能力,我们选择了一种名为STAT3的TF,并使用来自三种细胞类型(GM12878,CD4 + Th17和HeLa细胞)的ChIP测序数据训练了单独的模型来预测STAT3结合。结果表明,一些富集的5聚体在多种细胞类型之间共享,而另一些则具有高度的细胞类型特异性(图4)。
总而言之,这项研究提供了一种有价值的机器学习框架,可帮助解码TF结合其靶位点并可识别对结合作用最强的特定非编码核苷酸。这项技术未来的应用包括:新发现的TF和细胞类型,学习其他TF的细胞类型特定规则,研究TF结合区的选择信号,以及将核苷酸水平得分整合到GWASfine-mapping框架中等。
教授介绍

Melissa Gymrek,主要研究人类的遗传变异,特别是在基因的短串联重复序列中的遗传变异。她已获得一项专利的算法,该算法可从高通量测序数据中分析短串联重复序列,以进行遗传应用。她发明这项技术使此类研究得以扩展到整个人口规模,这有助于科学家了解这些小的基因片段如何导致不同人类特征的变化。
参考文献
1、An Zheng, Michael Lamkin, Hanqing Zhao et al. Deep neural networksidentify sequence context features predictive of transcription factor binding (2021). https://www.nature.com/articles/s42256-020-00282-y
发布于 02-01















 894
894











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









