







之前的教程里我们介绍了一个XDATCAR_toolkit.py工具,将VASP的AIMD的轨迹转成PDB的格式,再借用VMD和MD Analysis软件包分析RDF(径向分布函数)和RMSD(均方根偏差)。可惜的是,PDB文件不能承载速度信息,因此我们用c++重新写了一个VASP2GRO程序,输出可以记录原子位置和原子速度的GRO轨迹,它是 GROMACS 的私有文件格式,但是也被目前的大部分处理软件支持,比如可视化软件VMD。另外,VASP软件只输出了最后一帧的原子速度,因此无法得到与速度相关的性质,比如速度自相关函数等。我们借鉴樊哲勇老师博客中的方法(http://blog.sciencenet.cn/blog-3102863-1159419.html),通过向后差分的方法近似得到每一帧的原子速度。原理如下:
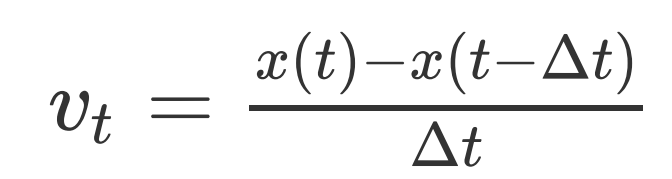
这样处理就舍去了第一帧的速度。如果想要得到比较精确的原子速度,可以通过牛顿定律求t时刻的速度vt:
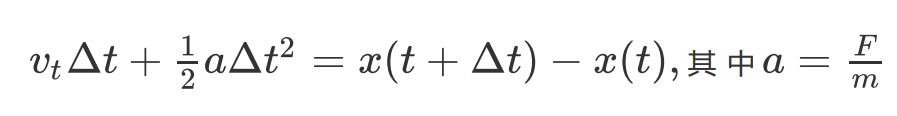
而不同时刻的原子受力,原子位置在OUTCAR中均有输出。本程序未采用该方法。VASP2GRO可以快速从OUTCAR中读取所需要的信息,并自动计算第2帧到最后一帧的原子位置和原子速度,输出到GROMACS的轨迹gro中。与之前的XDATCAR_toolkit.py一样,我们对周期性进行了处理,以保证计算得到的原子速度不会出现异常。
我们提供了编译好的VASP2GRO.exe和VASP2GRO_WIN.exe,分别为Linux和Window版本,可执行程序和其源代码可以在我的Github下载。(https://github.com/tamaswells/VASP_script/tree/master/VASP2GRO)。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章















 236
236











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









