






TCGA | GEO | 文献阅读 | 数据库 | 理论知识
R语言 | Bioconductor | 服务器与Linux

接前文:
转录组分析 | fastqc进行质控与结果解读
转录组分析 | 使用trim-galore去除低质量的reads和adaptor
转录组分析 | 使用Hisat2进行序列比对
转录组分析 | 使用SAMtools将SAM文件转换为BAM文件、排序、建立索引
转录组分析 | 使用RSeQC软件对生成的BAM文件进行质控
我们接下来使用Stringtie对数据进行下游处理。
一.StringTie介绍
StringTie 是用于 RNA-seq 的转录本组装和定量软件,StringTie 可以看做是cufflinks软件的升级版本,其功能和Cufflinks是一样的,包括下面两个主要功能:转录本组装和定量;相比Cuffinks, 其运行速度更快。该软件的官网:https://ccb.jhu.edu/software/stringtie/index.shtml。
1、Stringtie通过使用genome指导的组装的方法与从头组装的概念结合的新方法来改善转录组组装。
2、Stringtie的输入不仅可以是经过比对的结果,也可以是Stringtie通过从头组装read所得到的contig,当这两种输入都用到的时候,我们下面称之为stringtie+SR。
3、对于很多使用参考基因组辅助组装的方法,组装的的策略都是先对read进行cluter,然后建立一个graph model来推测每个基因所有可能的isoform,最终通过不同的graph的解析方法得到对转录本的组装结果。
4、有名的cufflinks用的是overlap graph,该模型中nodes代表fragment,如果两个fragment存在overlap并存在兼容的剪切模式,则对应的node连接起来。其解析方法为一种保守的算法,可以产生能够解释所有read的最少的转录本,尽管这种方法很吸引人,但是它没有考虑到转录本的丰度并且对于某些isoform来说该方法没有办法组装!
5、stringtie采用了组装转录本和估计表达量同步进行的方法,这不同于cufflinks的先组装后定量的策略。
6、首先,stringtie将read聚成cluster,然后采用了splice graph,其中node代表外显子或外显子的一部分,path将graph中可能 的剪切现象都连起来,最终对每个转录本通过创建一个网络流的方法,利用最大流算法(maximum flow algorithm)估计转录本的表达量。
7、最大流的问题是最优理论中的经典问题,但是目前还没有应用到转录本定量中。
8、与其它组装软件相比,stringtie具有很高的准确性和新型isoform的发现能力,其优势在于使用了网络流算法,同时stringtie也支持将read从头组装成更长的片段,这进一步提高了其组装的正确性。
9、其另一个优势在于它的最优化策略,它平衡了每次组装中每条转录本的覆盖度,这样可以对组装算法产生一定的限制,因为在组装基因组时,覆盖度是很重要的一个参数因为它需要被用来限制算法,否则组装器可能将重复的片段错误地堆叠到一起,相似地转录组装也是如此,在isoform中的每一个外显子需要有相似的覆盖度,如果忽略这个参数可能会产生一些保守但是错误的转录本,其中含有大量剪切位点的基因组装起来尤其困难。
常用的参数及描述:

-o []<out.gtf> #设置StringTie组装转录本的输出GTF文件的路径和文件名。此处可指定完整路径,在这种情况下,将根据需要创建目录。默认情况下,StringTie将GTF写入标准输出。-p <int> #指定组装转录本的线程数(CPU)。默认值是1-G #使用参考注释基因文件指导组装过程,格式GTF/GFF3。输出文件中既包含已知表达的转录本,也包含新的转录本。选项-B,-b,-e,-C需要此选项(详情如下)-l #将设置为输出转录本名称的前缀。默认:STRG-A #输出基因丰度的文件(制表符分隔格式)-C #输出所有转录本对应的reads覆盖度的文件,此处的转录本是指参考注释基因文件中提供的转录本。(需要参数 -G).-B #应用该选项,则会输出Ballgown输入表文件(* .ctab),其中包含用-G选项给出的参考转录本的覆盖率数据。(有关这些文件的说明,请参阅Ballgown文档。)如果选项-o 给出输出转录文件的完整路径,则* .ctab文件与输出GTF文件在相同的目录下。-b #指定 *.ctab 文件的输出路径, 而非由-o选项指定的目录。注意: 建议在使用-B/-b选项中同时使用-e选项,除非StringTie GTF输出文件中仍需要新的转录本,-B和-b选一个使用就行。-e #限制reads比对的处理,仅估计和输出与用-G选项给出的参考转录本匹配的组装转录本。使用该选项,则会跳过处理与参考转录本不匹配的组装转录本,这将大大的提升了处理速度。--merge #转录本合并模式。在合并模式下,StringTie将所有样品的GTF/GFF文件列表作为输入,并将这些转录本合并/组装成非冗余的转录本集合。这种模式被用于新的差异分析流程中,用以生成一个跨多个RNA-Seq样品的全局的、统一的转录本。如果提供了-G选项(参考注释基因组文件),则StringTie将从输入的GTF文件中将参考转录本组装到transfrags中。(个人理解:transfrags可能指的是拼接成更大的转录本片段,tanscript fragments)。在此模式下可以使用以下附加选项:-G #参考注释基因组文件(GTF/GFF3)-o #指定输出合并的GTF文件的路径和名称 (默认值:标准输出)-m #合并文件中,指定允许最小输入转录本的长度 (默认值: 50)-c #合并文件中,指定允许最低输入转录本的覆盖度(默认值: 0)-F #合并文件中,指定允许最低输入转录本的FPKM值 (默认值: 0)-T #合并文件中,指定允许最低输入转录本的TPM值 (默认值: 0)-f #minimum isoform fraction (默认值: 0.01)-i #合并后,保留含retained introns的转录本 (默认值: 除非有强有力的证据,否则不予保留)-l #输出转录本的名称前缀 (默认值: MSTRG)二.StringTie应用
1.单样本转录本组装
输入文档是BAM格式的比对结果文档,该文档必需经过排序。这些文档可以是来源于Tophat比对的结果文档,也可以是hisat2的结果文档经过转换和排序的文档(使用samtools)。我们前面使用的就是hisat2比对后用samtools排序后的bam文件。除此以外,我们还需要gtf注释文件。关于gtf注释文件格式参考文章:生信中常见的数据文件格式。
gtf注释文件可以去genecode下载你需要的gtf文件,我这里下载的是小鼠的。
genecode官网:https://www.gencodegenes.org/
我的gtf文件在/data/mouse_annotation/ 目录下:

单样本组装:
stringtie -p 8 -G /data/mouse_annotation/gencode.mouse.annotation.gtf -o cleandata/stringtiedata/CK-4.gtf cleandata/samtools_bam/CK-4_sort.bam批量处理:
for i in CK-7 CK-8 HGJ-10 HGJ-6 HGJ-9;do stringtie -p 8 -G /data/mouse_annotation/gencode.mouse.annotation.gtf -o cleandata/stringtiedata/${i}.gtf cleandata/samtools_bam/${i}_sort.bam;done查看输出结果:
ll -h cleandata/stringtiedata/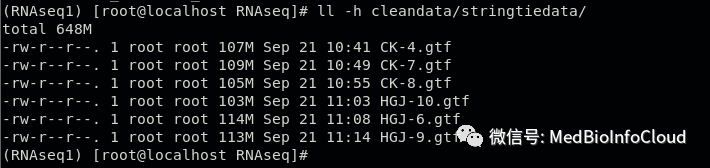
2.多样本转录本整合
stringtie --merge [options] gtf.list :转录组merge模式,在该模式下,Stringtie可以利用输入的一个gtf list并将他们中的转录本进行非冗余的整理。可以在处理多个RNA-seq样本的时候,由于转录组存在时空特异性,可以将每个样本各自的转录组进行非冗余的整合,如果-G提供了参考gtf文件,可以将其一起整合到一个文件中,最终输出成一个完整的gtf文件。
将文件名的完整路径输入到一个文件中:
find /data/RNAseq/cleandata/stringtiedata/ -name *.gtf > /data/RNAseq/cleandata/stringtiedata/merglist.txt查看一下merglist.txt文件中的内容:
cat /data/RNAseq/cleandata/stringtiedata/merglist.txt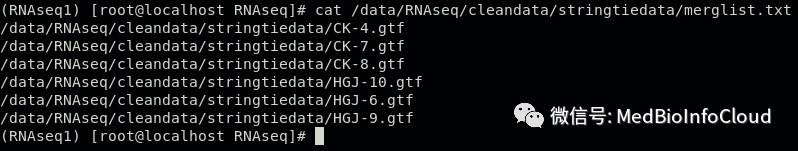
stringtie --merge -p 8 -G /data/mouse_annotation/gencode.mouse.annotation.gtf -o cleandata/stringtiedata/stringtie_merged.gtf cleandata/stringtiedata/merglist.txt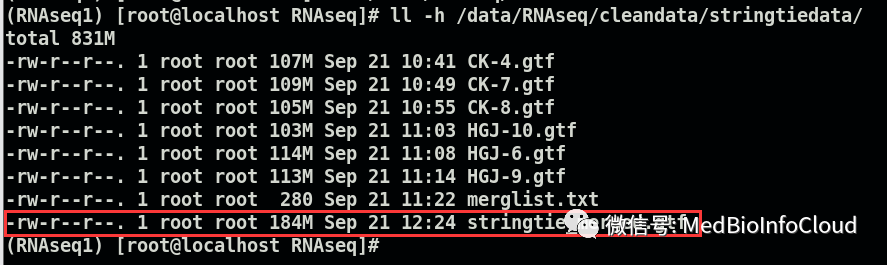
3. 使用gffcompare检验数据比对到基因组上的情况(可选)
程序gffcompare可用于比较、合并、注释和估计一个或多个GFF文件(“查询”文件)的准确性。
gffcompare [options]* {-i <input_gtf_list> | <input1.gtf> [<input2.gtf> .. <inputN.gtf>]}参数:
-i provide a text file with a list of (query) GTF files to process instead of expecting them as command-line arguments (useful when a large number of GTF files should be processed).-h/-help Prints the help message and exits-v/--version Prints the version number and exits-o All output files created by gffcompare will have this prefix (e.g. .loci, .tracking, etc.). If this option is not provided the default output prefix being used is: “gffcmp”-r An optional “reference” annotation GFF file. Each sample is matched against this file, and sample isoforms are tagged as overlapping, matching, or novel where appropriate. See the .refmap and .tmap output file descriptions below.-R If -r was specified, this option causes gffcompare to ignore reference transcripts that are not overlapped by any transcript in one of input1.gtf,…,inputN.gtf. Useful for ignoring annotated transcripts that are not present in your RNA-Seq samples and thus adjusting the “sensitivity” calculation in the accuracy report written in the file.-Q If -r was specified, this option causes gffcompare to ignore input transcripts that are not overlapped by any transcript in the reference. Useful for adjusting the “precision” calculation in the accuracy report written in the file.-M discard (ignore) single-exon transfrags and reference transcripts (i.e. consider only multi-exon transcripts)-N discard (ignore) single-exon reference transcripts; single-exon transfrags are still considered, but they will never find an exact match-D discard "duplicate" (redundant) query transfrags (i.e. those with the same intron chain) within a single sample (and thus disable "annotation" mode)-s path to genome sequences (optional); this will enable the "repeat" ('r') classcode assessment; should be a full path to a multi-FASTA file, preferrably indexed with samtools faidx; repeats must be soft-masked (lower case) in the genomic sequence-e Maximum distance (range) allowed from free ends of terminal exons of reference transcripts when assessing exon accuracy. By default, this is 100.-d Maximum distance (range) for grouping transcript start sites, by default 100.-p The name prefix to use for consensus/combined transcripts in the .combined.gtf file (default: 'TCONS')-C Discard the “contained” transfrags from the .combined.gtf output. By default, without this option, gffcompare writes in that file isoforms that were found to be fully contained/covered (with the same compatible intron structure) by other transfrags in the same locus, with the attribute “contained_in” showing the first container transfrag found. (Note: this behavior is the opposite of Cuffcompare's -C option).-A Like -C but will not discard intron-redundant transfrags if they start on a different 5' exon (keep alternate transcript start sites)-X Like -C but also discard contained transfrags if transfrag ends stick out within the container's introns-K For -C/-A/-X, do NOT discard any redundant transfrag matching a reference-T Do not generate .tmap and .refmap files for each input file-V Gffcompare is a little more verbose about what it's doing, printing messages to stderr, and it will also show warning messages about any inconsistencies or potential issues found while reading the given GFF file(s).--debug Enables debug mode, which enables -V and generates additional files: .Q_discarded.lst, .missed_introns.gtf and .R_missed.lst参考:http://ccb.jhu.edu/software/stringtie/gffcompare.shtml
我们将前面我们整合的转录本文件检验数据比对到基因组上的情况
gffcompare -r /data/mouse_annotation/gencode.mouse.annotation.gtf -G cleandata/stringtiedata/stringtie_merged.gtf会在输入的文件所在文件夹下产生几个文件:
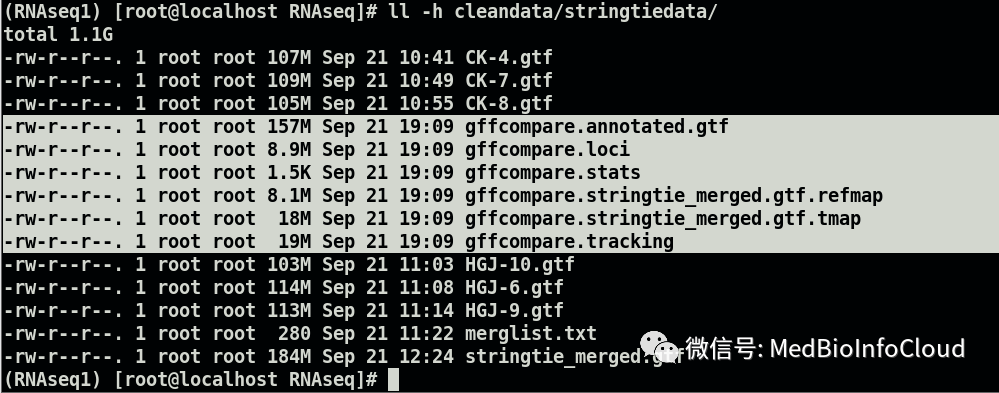
其中gffcompare.annotated.gtf存储的是StringTie组装的转录本与注释文件内的转录本的差别信息,通过class_code来表示:
head cleandata/stringtiedata/gffcompare.annotated.gtf |grep class_code | cut -d ";" -f 5-8
(RNAseq1) [root@localhost RNAseq]# head cleandata/stringtiedata/gffcompare.annotated.gtf |grep class_code | cut -d ";" -f 5-8 ref_gene_id "ENSMUSG00000116111.1"; cmp_ref "ENSMUST00000229069.1"; class_code "="; tss_id "TSS1" ref_gene_id "ENSMUSG00000115879.1"; cmp_ref "ENSMUST00000229227.1"; class_code "="; tss_id "TSS2" ref_gene_id "ENSMUSG00000116214.1"; contained_in "ENSMUST00000229087.1"; cmp_ref "ENSMUST00000229989.1"; class_code "="对照表如下:

gffcmp.stats文件存储比对结果的准确性和预测率。
cat cleandata/stringtiedata/gffcompare.stats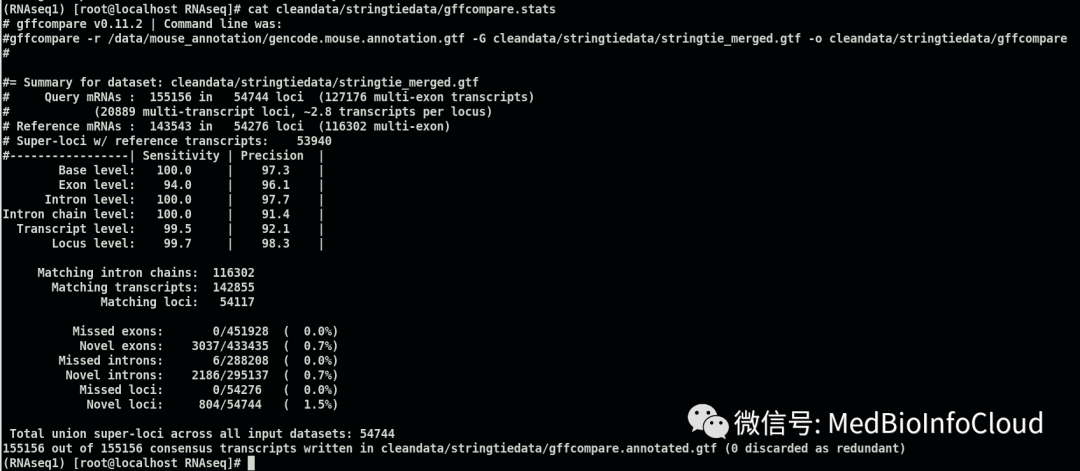
# gffcompare v0.11.2 | Command line was:#gffcompare -r /data/mouse_annotation/gencode.mouse.annotation.gtf -G cleandata/stringtiedata/stringtie_merged.gtf -o cleandata/stringtiedata/gffcompare##= Summary for dataset: cleandata/stringtiedata/stringtie_merged.gtf # Query mRNAs : 155156 in 54744 loci (127176 multi-exon transcripts)# (20889 multi-transcript loci, ~2.8 transcripts per locus)# Reference mRNAs : 143543 in 54276 loci (116302 multi-exon)# Super-loci w/ reference transcripts: 53940#-----------------| Sensitivity | Precision | Base level: 100.0 | 97.3 | Exon level: 94.0 | 96.1 | Intron level: 100.0 | 97.7 |Intron chain level: 100.0 | 91.4 | Transcript level: 99.5 | 92.1 | Locus level: 99.7 | 98.3 | Matching intron chains: 116302 Matching transcripts: 142855 Matching loci: 54117 Missed exons: 0/451928 ( 0.0%) Novel exons: 3037/433435 ( 0.7%) Missed introns: 6/288208 ( 0.0%) Novel introns: 2186/295137 ( 0.7%) Missed loci: 0/54276 ( 0.0%) Novel loci: 804/54744 ( 1.5%) Total union super-loci across all input datasets: 54744 155156 out of 155156 consensus transcripts written in cleandata/stringtiedata/gffcompare.annotated.gtf (0 discarded as redundant)4.重新组装转录本并估算基因表达丰度
利用merge得到的gtf重新对各个样本做定量,并创建ballgown可读取文件。
mkdir cleandata/ballgownfor i in CK-4 CK-7 CK-8 HGJ-10 HGJ-6 HGJ-9; do stringtie -e -B -p 8 -G cleandata/stringtiedata/stringtie_merged.gtf -o cleandata/ballgown/${i}/${i}.gtf cleandata/samtools_bam/${i}_sort.bam; done查看ballgown目录下生成的文件:
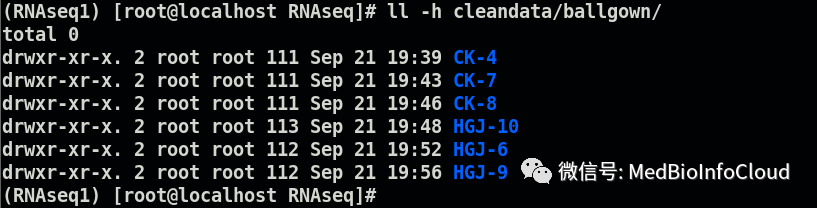
每一个样本对应一个文件夹。
查看其中一个:
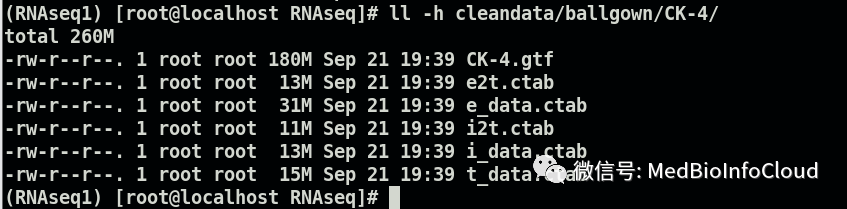
5.read count数据输出
这里需要prepDE.py这个脚本。
prepDE.py -i cleandata/ballgown/会在当前文件夹下产生2个csv文件。
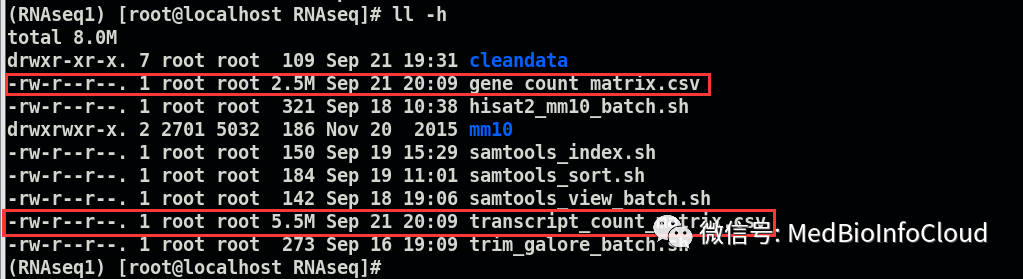
分别查看一下:
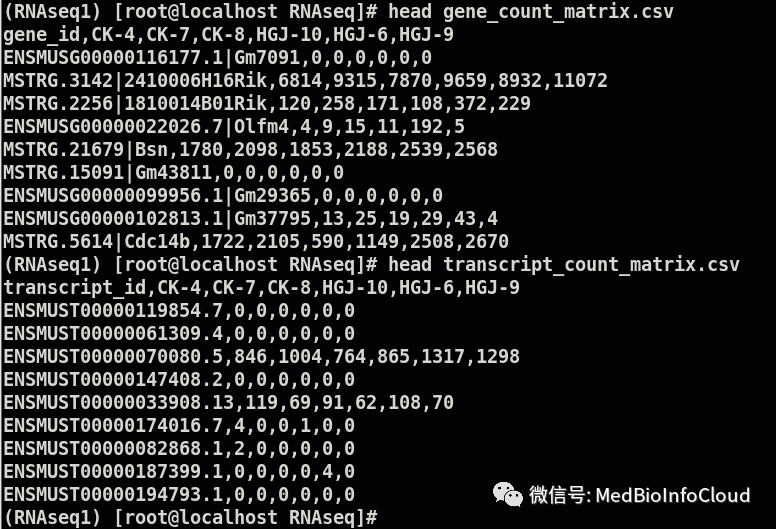
基础的转录组分析大概流程就是这样,当然,分析流程很多,我这里只是其中一个分析流程,可以参考文献,这里面涉及的软件也要不断应用,才知道那里出问题怎么解决,后续的分析算是下游分析,比如利用edgR包或DESeq2进行差异表达分析,这里得到的gene_count_matrix.csv数据就可以用于后续的下游分析了,比如差异表达分析,可参考TCGA序列的文章【TCGA】,本公众号文章目录可通过文章浏览:公众号文章目录。

















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









