





功能富集分析,是指借助各类数据库和分析工具进行统计分析。挖掘在数据库中与我们研究的生物学问题具有显著相关性的功能类别,期望发现在生物学过程中起关键作用的生物通路,从而揭示和理解生物学过程的基本分子机制。其中GO富集分析就是重要的一个内容。
通常我们可以很容易的通过各类在线网站得到GO富集结果,而后期的可视化往往是八仙过海,各显神通。
今天小鹿特别奉上一件法宝--GOplot,让你的可视化呈现与众不同。作为一款风格别致的GO可视化利器,这款R包相信大多数人已经有所接触,但很少去使用。接下来就带大家看一看GOplot都有哪些功能吧!
话不多说,首先简单欣赏一下GOplot例图:
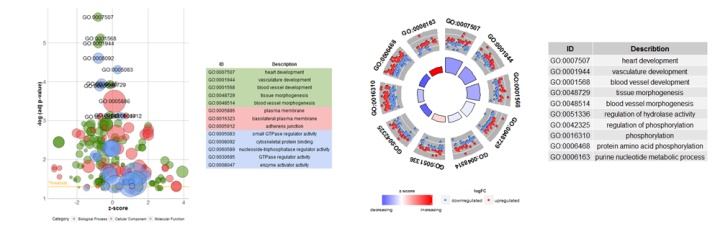
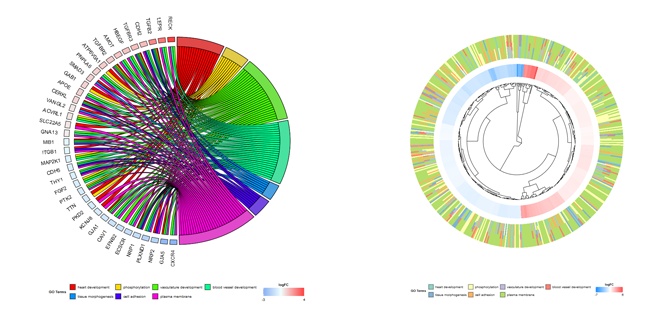
是不是比常规的柱状图和气泡图更加夺目呢?下面我们用第三幅为例,详细展示绘制过程:
数据准备
很多推文都有介绍过GOplot软件包,但都只是运行一下示例数据、展示结果。具体运用到自己的数据还是有些难度的,因此第一道坎就是我们要准备什么样的数据。

以上是绘制该图必需数据,首先是GO的id、term信息、所属分类、校正后p值以及所含的蛋白/基因名。值得注意的是,多个蛋白/基因名必须是逗号分隔;其次是蛋白信息,即蛋白与相对应的FC值。
数据“微整形”及绘图
1.加载R包,没有则需安装:
>library(GOplot)
载入需要的程辑包:ggplot2
载入需要的程辑包:ggdendro
载入需要的程辑包:gridExtra
载入需要的程辑包:RColorBrewer
由依赖包可以看出,GOplot所绘制的图都是依据ggplot的。
2.导入数据
library(openxlsx)
go<-read.table("GO.xls",sep="t", header=T, quote="")
pro<-read.xlsx("protein.xlsx")
#如果go最后一列有引号,应使用gsub去掉
3.数据“微整”
colnames(go)<-c( 'ID', 'term','category','adj_pval','genes')
pro$FC<-log(pro$FC)
colnames(pro)<-c("ID","logFC")
如图所示:
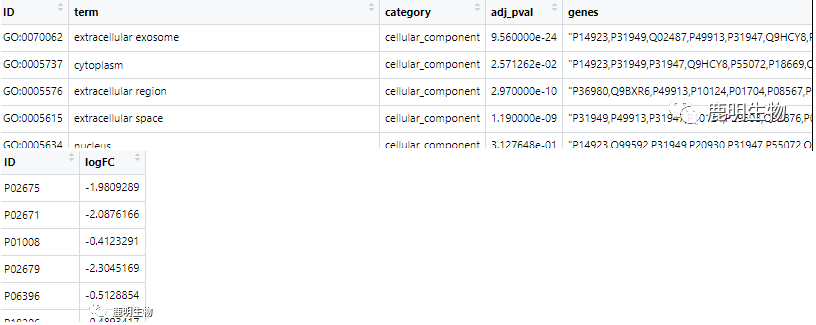
总的来说,就是换了列名,计算了logFC,至于原因;当然是因为该程辑包里面把名称固定了。
绘图前的“黑暗”
虽然距离绘图不远了,但图所对应的数据并不是微调后的两个数据框,我们还需伪装一下打入GOplot内部。伪装其实是对输入的两张表进行整合,把每个term所对应的蛋白和logFC逐个排列下来。
运行命令:
circ <- circle_dat(go,pro)
结果展示:
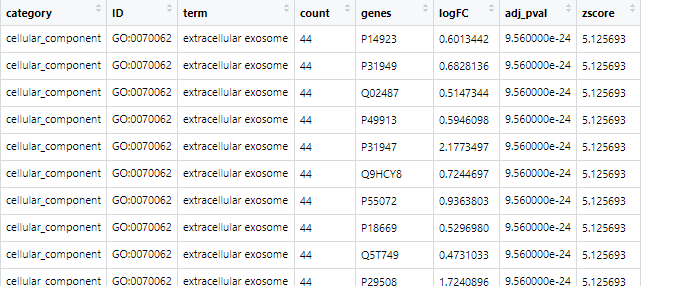
值得注意的是最后一列的zscore,公式为:
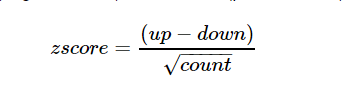
zscore反映该功能所含蛋白的总体表达趋势,大于0为上调,小于0为下调。
打入内部后,我们就要回想一下目的,和弦图主要是展示感兴趣的蛋白和感兴趣的GO条目之间的关系。那么先选定目标,以前六条为例,蛋白为所有蛋白,因为不在选定条目内的蛋白会被剔除。
gop<-go[1:6,]
chord <-chord_dat(data = circ, genes = pro,process = gop$te
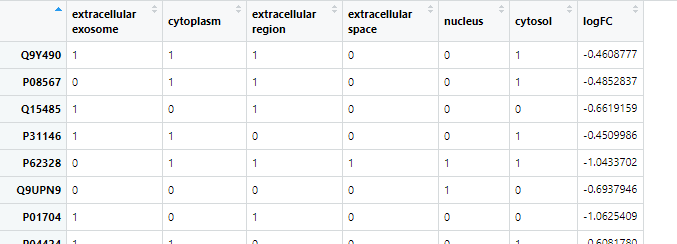
运行命令得到一个矩阵,表示每个蛋白是否具有选定的功能,具有显示为1,否则为0。
绘图!!!
运行代码:
GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 4)
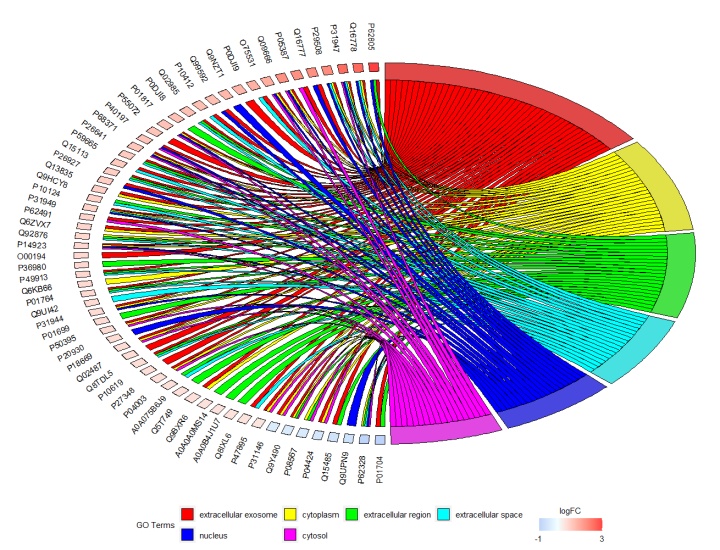
左面为蛋白名,红色表示上调,蓝色表示下调。右面为所选GO term。
私人订制
以上实现了如何用我们的数据绘制和弦图,要得到一张非常满意的个性化元素强烈的图,就是这篇文章的重点了,我们最直观感受到的是该图的颜色,所幸还有一个可以更改颜色的参数,即ribbon.col,输入命令:
GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 4 , ribbon.col=brewer.pal(6, "Set3"))
更改后如图:
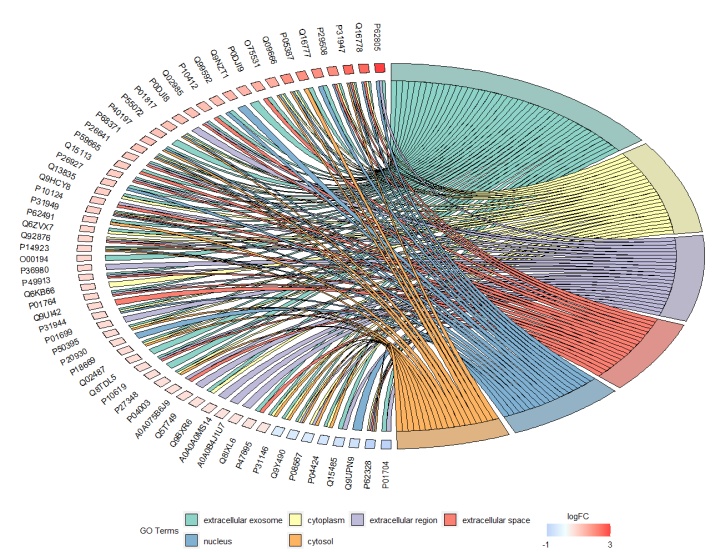
最后来关注一下图例部分,我们经常遇到条目名称较长的情况,这样的话会影响图例显示的美观度,可是修改的话,绘图函数里并没有提供这个参数,那该怎么办呢?
这时候就需要我们去深入研究一下这个函数了,直接输入函数名回车就可以看到整个代码,将代码复制下来修改后赋予另一个函数名再调用,就是私人订制的过程。
示例1:修改term图例排列方式—两列显示
①查看函数

②修改函数

由函数代码可以看出,默认是4列显示,两列的话将ncol=4改为ncol=2即可,修改后保存该函数为GOChord_new,结果如下:
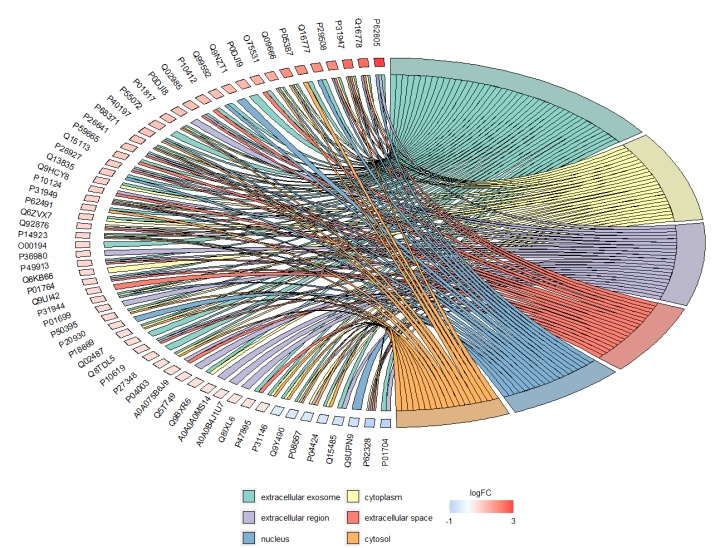
示例2:移动logFC图例位置—放到最底层中间位置
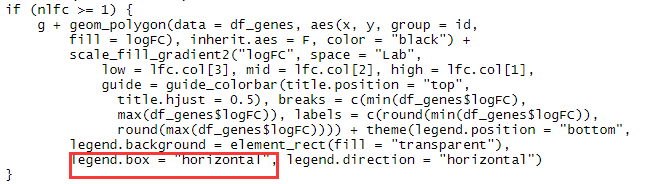
将legend.box = "horizontal"改为legend.box = "vertical"即可,结果如下:
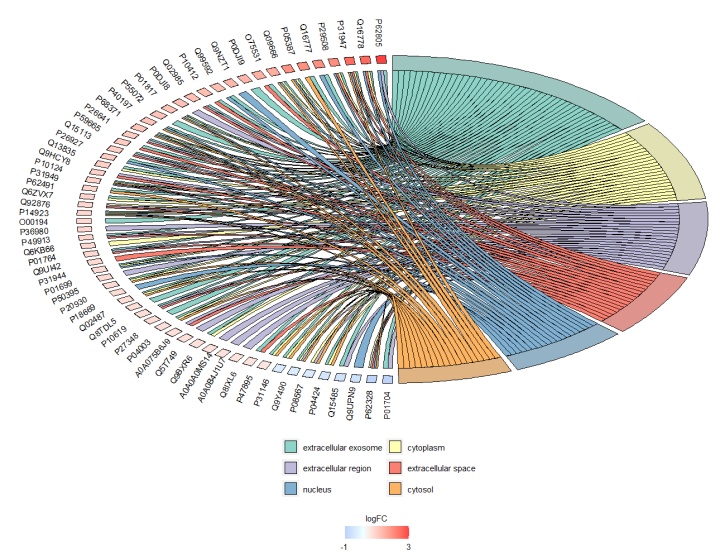
以上两个示例是修改图片风格的,当然我们也可以修改一些被固定的参数,比如前面数据准备里的分隔符和列名,都可以通过定制来使其更符合我们的习惯和要求。
总结
生信分析过程中,我们经常会遇到一些可视化效果很好但灵活度不够的R包,这就会让结果成为将就,但我们不愿将就!希望本期的这个R包不太冷——Goplot定制包,能为大家在使用R package时提供一个新思路。
猜你还想看
◆生信教程:10行代码让你的相关性图貌美如花
◆生信教程:对话百年名画--文章绘图配色高级又简单!
◆生信教程:只需3分钟Get“代谢通路分析神器”
◆生信教程:【指南】Cytoscape之stringAPP蛋白互作分析详解
END
文章来源于鹿明生物
















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









