







桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
123篇原创内容
公众号
今天开启单细胞之旅,单细胞其实我也算接触较早的项目,之前在老东家参与开发单细胞测序的实验开发以及后期生信流程的搭建,那时开发的技术还真的就是单个细胞的,搞了几项发明专利:
-
201610701005.6 用于单细胞全基因组中亚硫酸氢盐测序
-
201511029177.5 一种单细胞简化代表性重亚硫酸氢盐测序的方法及试剂盒
-
201510680615.8 构建高分辨率、大信息量单细胞Hi-C文库的方法
-
15906427.8 METHOD FOR CONSTRUCTING HIGH-RESOLUTION SINGLE CELL HI-C LIBRARY WITH A LOT OF INFORMATION
基于以上经验,有需要软著以及专利发表的老师可以联系桓峰基因合作,我们真诚期待与各位老师合作搞科研,重知识,重产出,走产学研一体化的科研理念!
前 言
单细胞测序技术,简单来说,就是在单个细胞水平上,对基因组、转录组及表观基因组水平进行测序分析的技术。传统的测序,是在多细胞基础上进行的,实际上得到的是一堆细胞中信号的均值,丢失了细胞异质性(细胞之间的差异)的信息。而单细胞测序技术能够检出混杂样品测序所无法得到的异质性信息,从而很好地解决了这一问题。
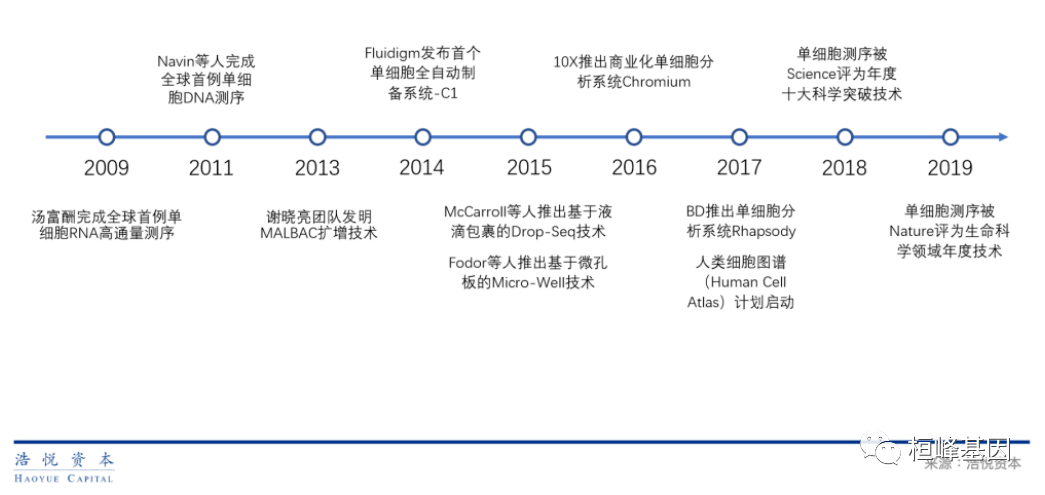
从单细胞测序的发展进程来看,我还真在2013年开发MALBAC测序开始参与的,那时我是单位的一块砖,哪里需要哪里搬,后来转去研究临床了,不过接触项目越多,越觉得蛮有意思的,现在又重拾单细胞这个块,更上技术发展的脚步!
目前普遍认知中的“单细胞测序”技术其实比较“年轻”,最早起源于2008-2009年汤富酬教授的研究成果,汤富酬教授于2009年完成了世界首例单细胞RNA测序技术开发及试验。但在2015年之前,受限于现有单细胞分离技术,早期单细胞测序技术仅能检测少量细胞,且成本高昂。到了2015年,随着基于细胞条形码标记技术的高通量(或"海量")单细胞分离与测序技术的成熟,科研人员有能力一次性对数千,甚至上万个单细胞进行标记和分析,使单细胞测序技术在临床上应用成为可能。
10X Genomics测序原理
10X Genomics已经广泛应用于单细胞测序、组装领域,现在也是火得不行。
产品网站有介绍的很清晰,Products - 10x Genomics
https://www.10xgenomics.com/products
我们看下产品都有什么,后续都会涉及到哪些测序类型?从单细胞的定量基因表达到全转录组,再到空间染色体与表达结合测序,最后就是单细胞捕获测序,非常全面的产品!


基本原理是通过将来自相同DNA片段(10-100kb)的reads加上相同的barcode,然后在illumina平台上进行测序,从而实现长片段的测序。其基本原理是同一长片段的reads会具有同样的标签,称为linked-reads,利用这些barcode的信息,可将短reads拼接为长reads。这样的linked-reads可进行结构变异检测及单倍型定相的分析。
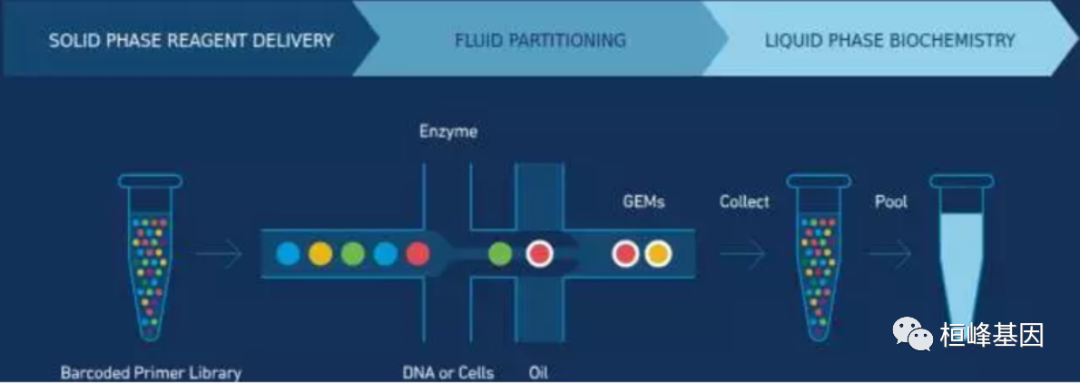
总结:10X Genomics发明了一种独特的barcode建库技术,能将同一种barcode的reads连成linked reads从而提高了reads的长度。但10X Genomics的测序之本仍然是illumina,GC偏向性是硬伤。同时barcode技术有一定的环境限制。
单细胞测序类型
单细胞测序在组学上包括基因组,转录组,甲基化,蛋白以及空间位置等表观组学的测序以及生信分析,而测序一般都是使用10x Genomics 测序。
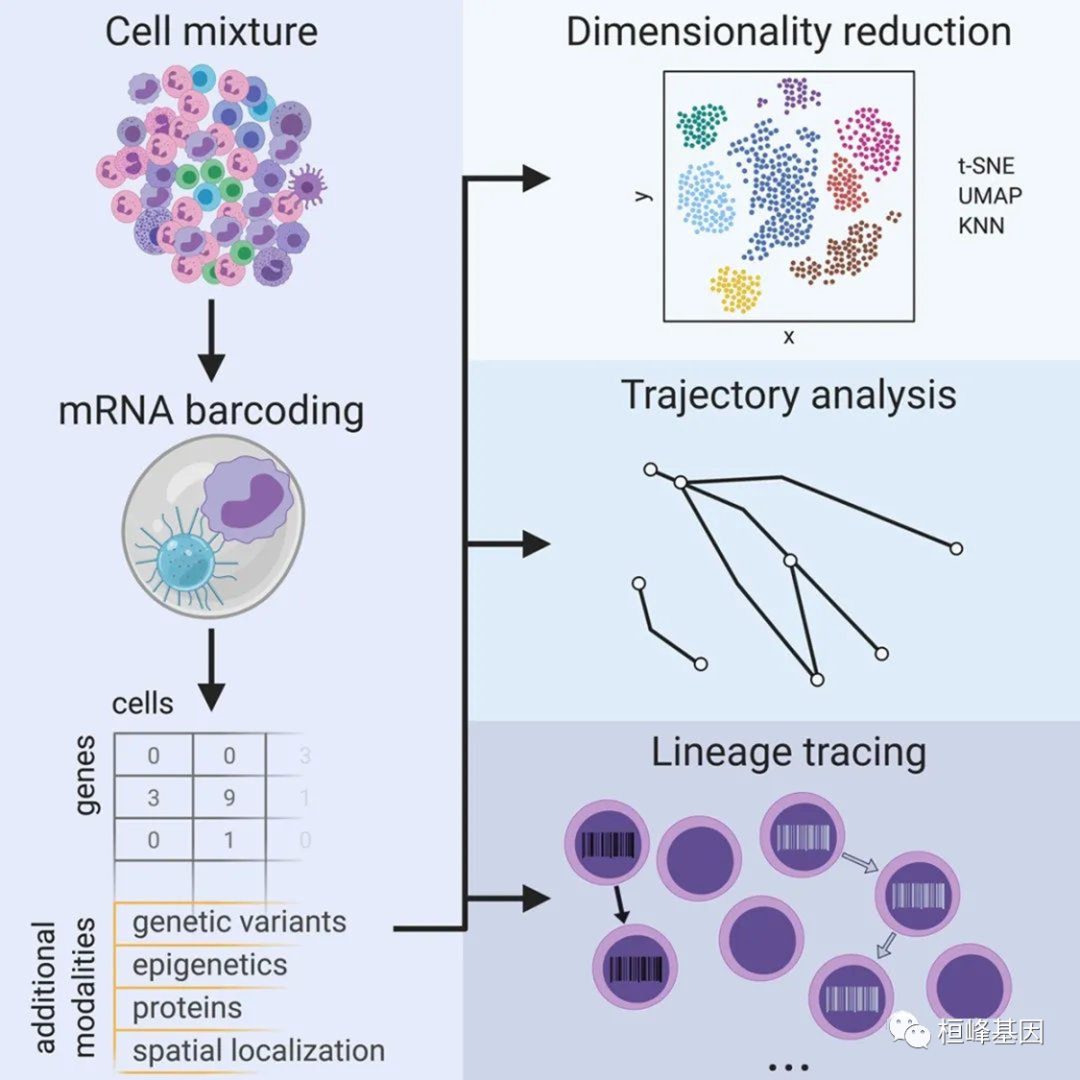
- 单细胞转录组测序
测量单细胞的转录组仅仅出现几年,但它已经成为一种非常受欢迎的检测方法。通过单细胞RNA-seq,实现了研究单个样本中细胞类型多样性的能力。在不同的细胞类型中,无偏和可复制的基因表达模式需要大量的细胞。技术的发展和改进推动了单细胞RNA-seq分析中可研究的细胞数量的持续和指数增长。从单细胞RNA-seq实验获得的数据中实现这种增长的关键技术发展。从上图可以看出,2010-2014年之间,单细胞测序领域采用的主流技术的细胞通量都在100以内,2015年后,随着海量单细胞分离技术的出现,取得了高速的进步;进行单细胞测序前,首先需要分离单个的细胞,如何低成本的获得大量的单个细胞是单细胞测序流程中最重要的环节。2015年技术大爆发后,目前市场上比较成熟的单细胞分离方法主要是10X Genomics的Chromium(“液滴法”)及BD的Rhapsody(微孔法)平台。
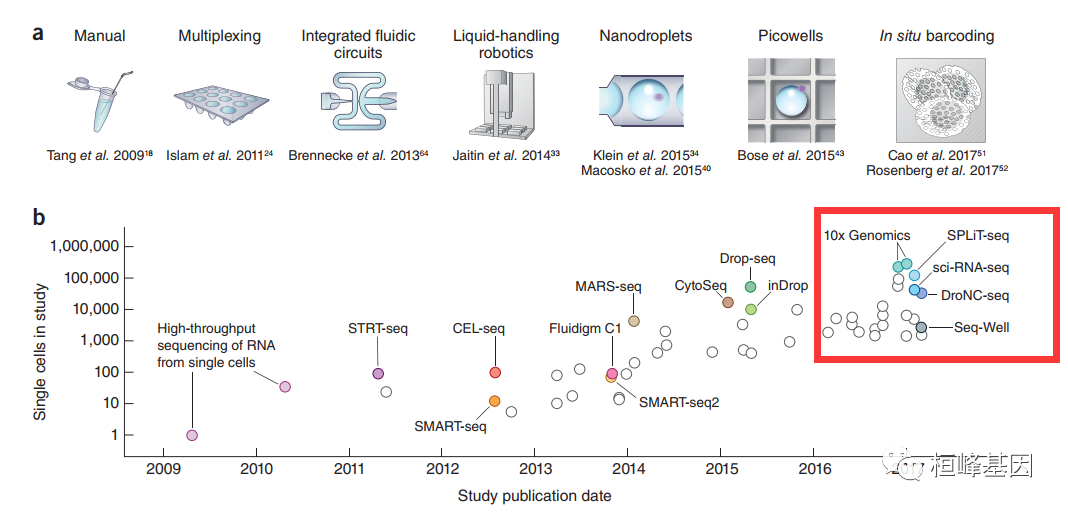
单细胞转录组测序得从这篇文章说起,根据文章中的测序图解我们来浅显的了解一下测序原理,方便我们后续生信分析时出现的问题可以快速的分析导致出问题的原因,到底是实验方法上还是生信分析的方法上,搞清最基础的数据来源,我们才能够得心应手。

我们通过文章中fig1来理解其实验及测序原理,从复杂的组织中分离细胞,并进行单细胞转录组建库,合成唯一识别码(UMIs),最后完成建库!
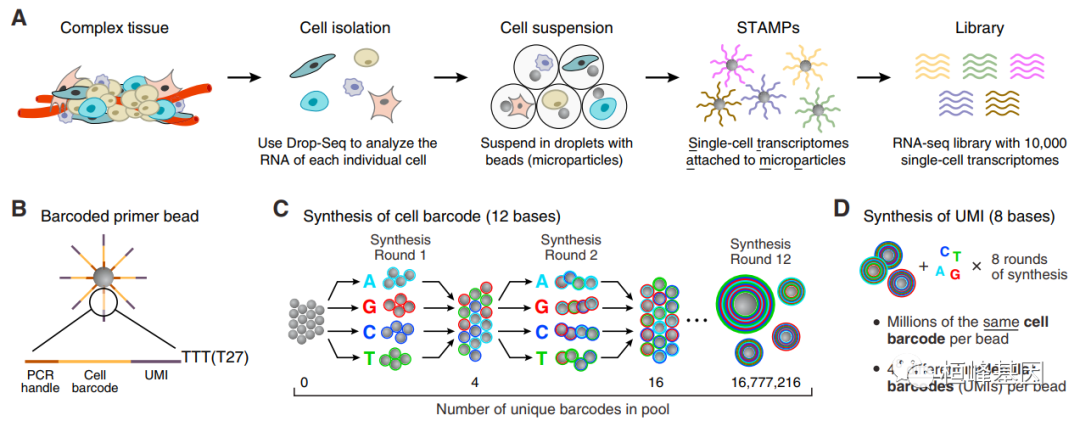
(A) Drop-Seq 条形码示意图。一个复杂的组织被分解成单个的细胞,然后这些细胞和微粒一起被包裹在液滴中(灰色圆圈)传递条形码引物。每个细胞都在液滴中分解;信使 RNA 与同伴微粒上的引物结合。信使 RNA 是逆转录成cDNA,产生一组珠子,称为“附着在微粒上的单细胞转录组”(STAMPs)。条形码在池中进行扩增,用于高通量 mRNA-seq 分析任意数量的单个细胞。
(B) 微粒上引物序列。所有珠上的引物都包含一个共同的序列 (“PCR handle ”),以便在 STAMPs 后进行 PCR 扩增形成。每个微粒包含超过108个单独的引物,它们共享相同的“细胞条形码” ©,但有不同的独特分子标识符(UMIs),使mRNA转录本计数 (D)。所有捕获mRNA的引物序列的末端都存在一个30 bp的 oligo dT 序列。
© 细胞条形码的分裂池合成。为了生成细胞条形码,微粒子池被重复地分裂成四个相同大小的寡核苷酸将四种 DNA 碱基中的一种加入到合成反应中,然后在每个循环后聚合在一起,共 12 个拆分池循环。条形码在任何单个珠上的合成反应反映了该珠通过一系列合成反应的独特路径。其结果是每一个都形成了一池微粒在其全部引物序列中拥有 412(16,777,216) 个可能序列中的一个。
(D) Unique Molecular Identifier (UMI) 的合成。在“分-池”合成循环完成后,所有的微粒一起8轮合成,每个循环中所有4个 DNA 碱基都可用,这样每个引物可以从48(65,536)个碱基中获得一个 UMI。
**单细胞表达谱测序与bulk RNA测序对比。**传统的二代测序最为人熟知的就要数RNA-seq了。RNA-seq是提取组织、器官或一群细胞的混合RNA(bulk RNA)进行测序,能够得到的是一群细胞的转录组的平均数据,细胞群体中单个细胞的特异性信息往往被掩盖(比如特异表达的基因或RNA不同的剪接体)。而随着对生物结构功能的深入研究,人们越来越清楚地认识到,哪怕看似相同的细胞群,细胞之间的转录组表达水平也是存在差异的。以肿瘤为例,肿瘤中心的细胞,肿块边缘的细胞和肿块周围的细胞,乃至远端转移的细胞,其转录组等遗传信息一定是存在差异的,而传统的研究手段通常将整个肿块整体进行研究,或者将肿块简单分区分割,得到每一部分细胞基因表达的平均值,丢失了每个细胞的异质性信息,使科研人员对肿瘤微环境中各种细胞转录组表达及免疫功能的理解和认识始终无法深入。由此可见,为了得到原始和真实的细胞遗传信息,对转录组的分析须在单细胞水平上展开。单细胞表达谱测序技术可以从混杂组织中捕获单个细胞,并能从中获取单个细胞的表达信息,检测出混杂样品中细胞的异质性数据。
- 单细胞全基因组测序
根据分子生物学的中心法则,如果说转录组表达量的高低是“结果”,那么要想找到导致这种结果的“原因”,我们需要在基因组的层面上进行研究。众所周知,单细胞转录组测序大大提高了我们研究的分辨率。但是单细胞基因组测序,依然是困难重重。其核心痛点在于如何将单个细胞核内极其微量的 DNA 高保真、高均一和高产率地扩增至可测序水平,从而实现在单个细胞水平对多种基因变异类型 (包括极具挑战的单细胞 SNV 和 CNV 检测等) 进行高清分析的目的,满足辅助生殖、遗传病、肿瘤学、神经学、细胞学和感染病等研究领域的前沿应用需求。
从方法学角度来看,获得高覆盖率高保真性的全基因组扩增产物是准确全面的测序结果的保障。多重置换扩增(multiple displacement amplification,MDA)利用随机引物和等温扩增可以获得高保真的DNA大片段,但该方法的主要缺陷在于非平衡的基因组覆盖率、扩增偏倚、嵌合序列及非特异扩增等。尽管各种改进的策略正在逐步减少这些缺陷,高覆盖率、高保真性及高特异性的扩增仍然是亟待解决的问题。另外,还有科研人员利用DOP-PCR进行全基因组扩增(whole-genomeamplification,WGA)及DNA测序对单个乳腺癌细胞进行了拷贝数变异的分析,进而推断出细胞的群体结构和肿瘤的进化过程。但是由于该方法的基因覆盖率较低,而且不能在单个核苷酸的分辨率上评价单个肿瘤细胞的遗传学特征,故并不能检测在肿瘤发展过程中发挥重要作用的单个核苷酸的改变。2012年,哈佛大学谢晓亮院士在《Science》发表了单细胞全基因组扩增新技术MALBAC(Multiple Annealing and Looping Based Amplification Cycles,简称MALBAC),即多次退火环状循环扩增技术。不同于以往的非线性或指数型****扩增方法,MALBAC技术利用特殊引物,使得扩增子的结尾互补而成环,从而很大程度上防止了DNA的指数性扩增,从而解决了基因组扩增对微量初始模板过大的扩增偏倚,并使基因组测序的模板需求量从µg级降至单细胞水平。MALBAC技术原理如下:
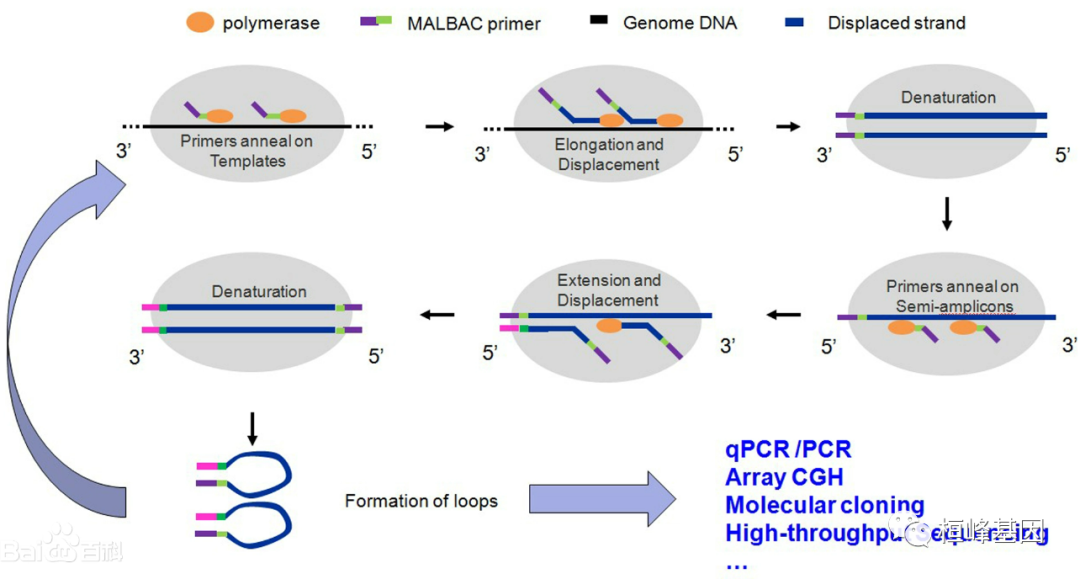
MALBAC引物的共有序列为27 ntps,后面有8个随机核苷酸,将引物退火到基因组DNA模板上。链置换合成产生部分扩增子,随后在94 c位点从模板变性到基因组DNA模板上的新位置产生更多的部分扩增子,这增加了基因组的覆盖率,从而降低了扩增偏差。在部分扩增子上进行引物和延伸,得到的扩增子在第5端具有MALBAC引物序列,在第3端具有互补序列。94℃变性可以再生出原来的模板和一个更大、更多样化的部分扩增细胞池。全扩增形成环,可能对后续的扩增和杂交有抗性。8个周期产生完整的扩增子,使用与MALBAC引物公共区域互补的引物,指数扩增约14-21个周期。
临床上的应用
精准医学技术发展至今,已经在基因检测领域取得了一定建树:目前广泛使用的NGS(二代测序)技术能够灵敏、准确的发现人体组织、甚至血液中游离的DNA的突变结果,并对治疗进行指导。但长期以来,针对人类基因组和疾病的研究都是基于“组织”或“血液”这样的整体概念,未能从单个细胞层面的基因表达,细胞行为的分辨率上进行分析。
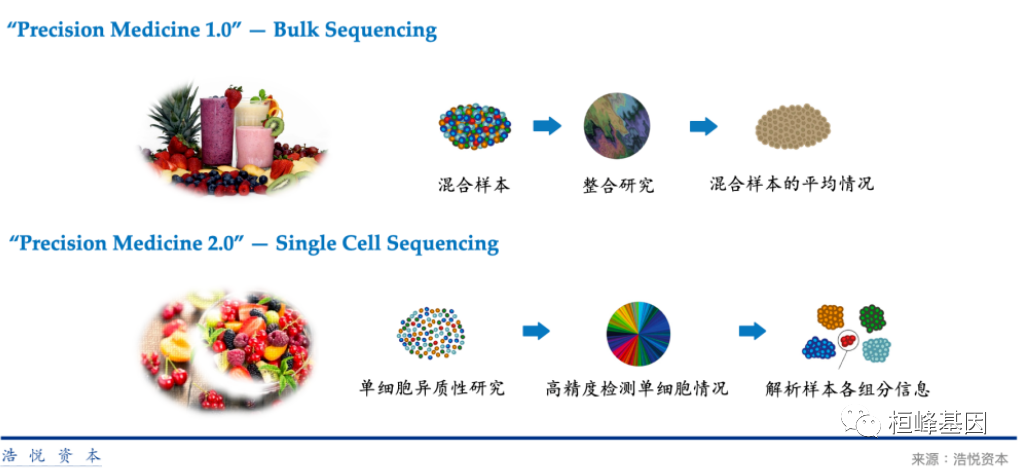
细胞是生物体结构和功能的基本单位,长期以来,针对人类基因组和疾病的研究都是基于“组织”或“血液”这样的整体概念,未能从单个细胞层面的基因表达,细胞行为的分辨率上进行分析。目前我们熟悉的二代测序检测结果,仅能反映一群细胞中平均化的各类信号表达的中值或“主流”信息,单个细胞层面的“个体差异”信息则被忽视。对于肿瘤研究、神经疾病、免疫疾病等涉及细胞间异质性的疾病研究,从单细胞层面分析细胞基因表达,细胞间互作等是有重大意义的,因此能够解释单个细胞基因结构与表达状态,反应细胞间异质性的单细胞测序技术应运而生。单细胞测序技术与传统测序比较,就像“水果”和“混合果汁”的对比,可通过更高分辨率的单细胞层面发现并解析各组分之间的具体信息及关联性,因此可以看作是“Precision Medicine 2.0"。
单细胞技术在国内外成为了热门科研领域研究的技术手段新宠。随着精准治疗时代的到来,未来单细胞技术,将会有助于人类的科学研究以及临床应用,尤其是在肿瘤、微生物、神经科学、免疫学等领域,应用会越来越广泛。
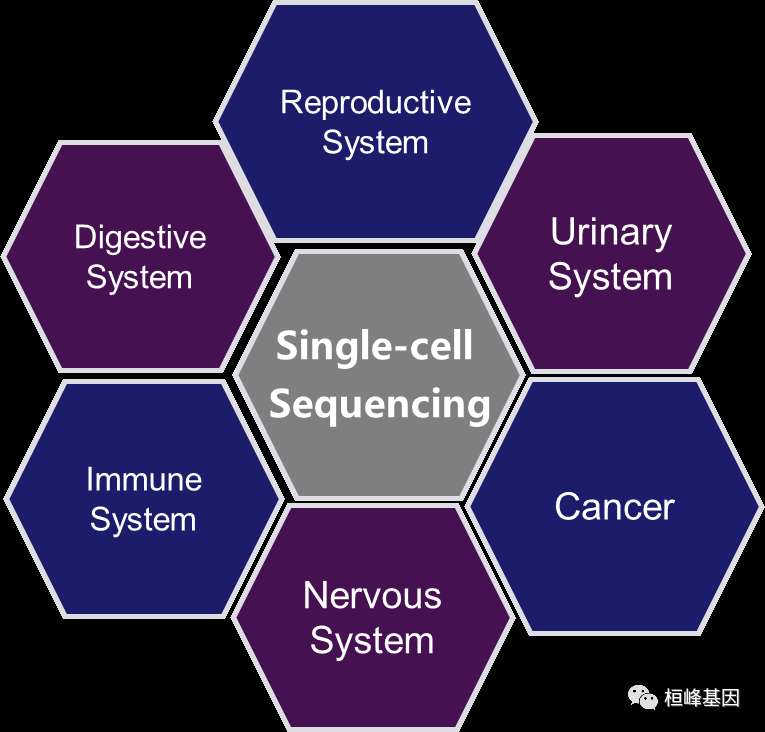
**肿瘤研究:**肿瘤细胞的突变速率非常快,而且肿瘤组织是一种高度异质性的组织。确定肿瘤组织中存在哪些细胞亚群(或者叫克隆)具备转移能力,哪些克隆对化疗药物是敏感的,这些信息对于临床工作非常有帮助。结合近年来的研究进展,可以看到单细胞测序对于肿瘤免疫治疗等新手段可以提供巨大帮助,比如发现/确认新靶点等。新格元与上海肺科医院等单位联合开展的中国人群肺腺癌、鳞癌免疫基因组计划致力于考察肺癌肿瘤微环境中不同类型细胞的组分和特征,以及单细胞表达谱和疗效关系。
**自体免疫疾病:**人体内的免疫细胞为了实现不同功能,具有极强的杂合性,单细胞测序能在测定每一类细胞的基因表达的同时,对这些细胞进行区分,甚至进行更准确的亚型分类,为免疫学研究和相关疾病的治疗提供帮助。
**神经生物学:**单细胞测序能够比较好的研究单个细胞的表达谱,对神经细胞进行种类细分,并对细胞间的通信提供更多的信息,从而解开神经信号传递的秘密,为疾病治疗提供更多的思路。
**发育研究:**人体内的各类组织器官的发育都会受到细胞微环境的影响,不同个体的细胞发育命运各不相同。单细胞测序可以帮助我们对发育过程中的每个细胞个体的基因表达、作用以及细胞间的通信和影响进行解释。
- 单细胞测序的技术将彻底改变整个生物科学
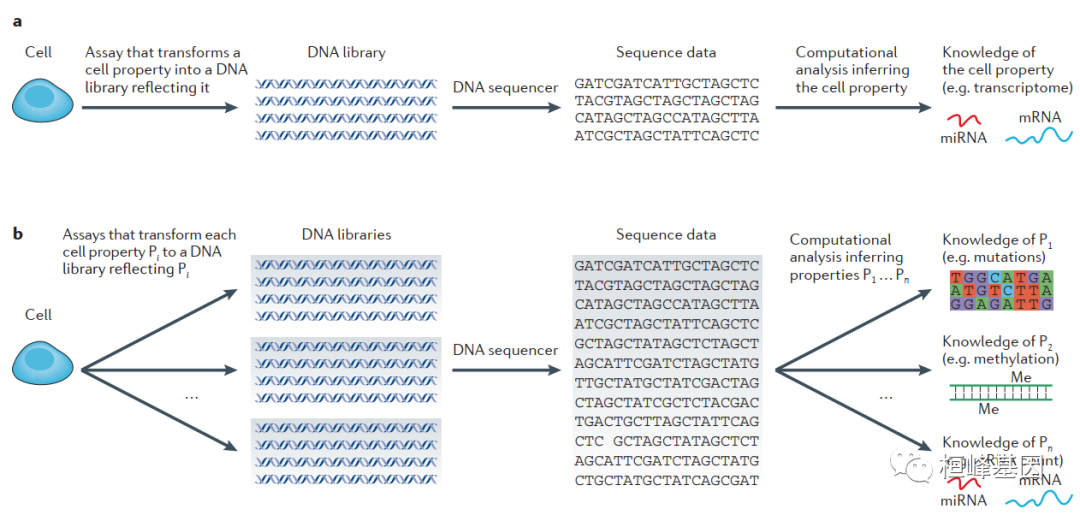
基于单细胞测序的分析方法及其预期整合。目前的实施包括单细胞基因组学(靶向外显子组或突变分析,拷贝数变异和生殖细胞中的重组分析,转录组学(转录组分析和免疫系统中的重组分析)和表观基因组学。我们预计在十年内,综合单细胞DNA测序分析体系结构将允许同时分析单个细胞的多种属性,包括基因组表观基因组学(甲基化,染色质和构象分析)、转录组学(转录组分析,等位基因特异性表达和分子计数)和蛋白质组学。
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,敬请期待!!
References:
-
Macosko EZ, Basu A, Satija R, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202-1214. doi:10.1016/j.cell.2015.05.002
-
Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13(4):599-604. doi:10.1038/nprot.2017.149
-
Yilmaz S, Singh AK.Single cell genome sequencing. Curr Opin Biotechnol 2012; 23(3): 437-43.
-
Zong CZ, Lu SJ,Chapman AR,XieXS.Genome-Wide Detection of Single Nucleotideand CopyNumber Variations of a Single Human Cell. Science 2012;338(6114):1622-6.
-
Lu SJ, Zong CZ, Xie XS, et al. ProbingMeiotic Recombination and Aneuploidy of Single Sperm Cells by Whole GenomeSequencing using MALBAC. Science 2012;338(6114):1627-30.
-
Balogh M K. Application of whole genomeamplification for forensic analysis. Elsevier 2006; 1288:725-727.
-
Pinard R, de Winter A, Sarkis G J, etal. Assessment of whole genome amplification-induced bias throughhigh-throughput, massively parallel whole genome sequencing. Bmc Genomics 2006;7(1): 216-20.














 902
902











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









