






一.从头预测方法
从头预测,来源于拉丁文ab initio或de novo,严格的讲,这两者是有区别的,前者ab initio意思是from tlIe beginning,指基于第一性原则(6rst principles)而不依靠同源序列、数据库、二级结构等其它信息,仅靠一条蛋白质序列产生三维结构。后者de novo意思是from the new,是一个更宽泛的含义,指不需要PDB中的同源模板而是依靠对其他结构的观察来预测。
从头预测方法具有一定的特点和优势,它适用于同源性小于25%的大多数蛋白质。
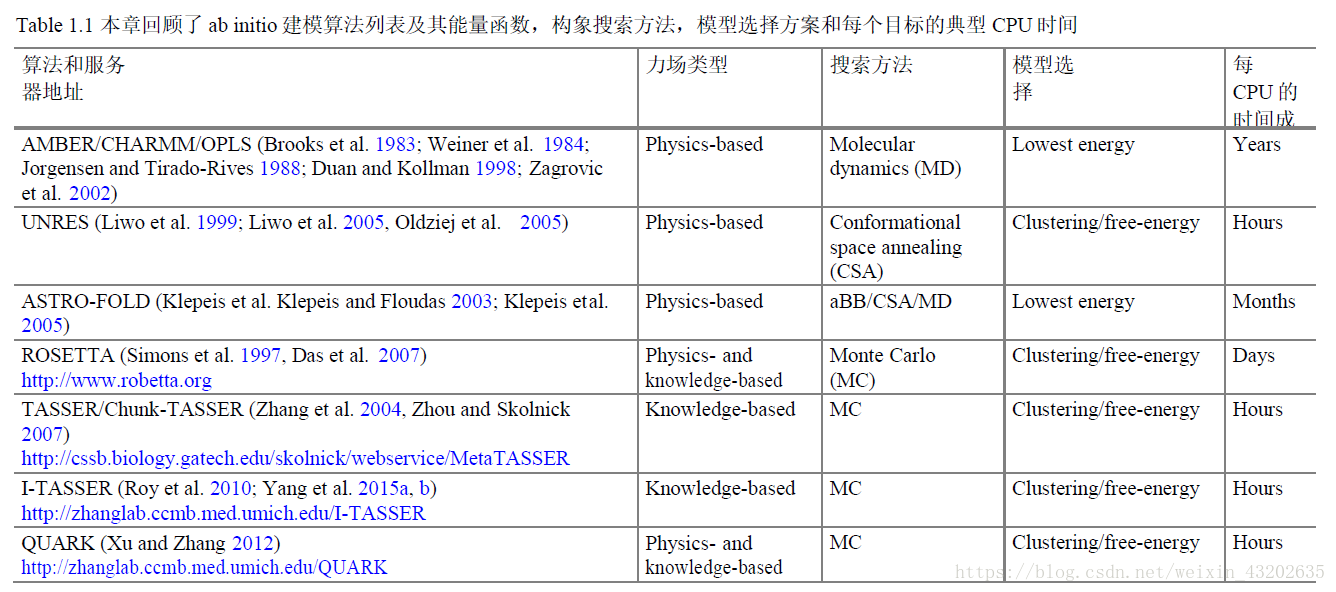
1. 能量函数建立
建立一个能区分蛋白质正确构象与其它构象的能量函数,是从头预测方法中关键性的第一步。理想的能量函数(又称打分函数,势能函数)应该能够精确表达蛋白质的所有原子空间位置及其能量之间的关系,通过能量极小化找到天然构象。
1.1基于物理经验的能量函数
物理学上又称分子动力学方法,是分析粒子之间相互作用的基本原理后得到的经验公式,它能反映客观存在于蛋白质内部或者蛋白质分子和溶剂分子之间的物理作用,尽管这类能量函数比较复杂且计算成本很高,它在计算高分辨率结构方面依然有很大的应用。
典型的经验公式包括键伸缩能、键角变形能、键的转动(二面角)能和非键相互作用,公示如下:
上面式中能量E是N个原子位置的函数。
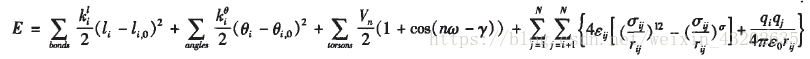
典型物理能量函数是Seheraga建立的联合残基力场(uNRES)。联合残基力场提供了蛋白质分子中原子间相互作用的能量函数,简化了模型,所以相对全原子力场,计算量较少,同时考虑了多种作用力形式,一定程度上保证了建模的精度。
1.2基于知识的能量函数
利用蛋白质结构数据库(PDB)中的已知结构数据作为学习样本,计算得到的具有统计性质的区分参数,然后根据能量按Bolzman分布的原理反推出一个能量函数。
通常,为得到这种经验性的能量函数表达式,我们首先要选择一系列已知结构的蛋白质,然后对每一个氨基酸采用统计学方法分析在三维空间上与其相邻的氨基酸,根据不同氨基酸的相对位置得出一个得分矩阵。
2. 构象搜索
能量函数确定之后,从头预测方法便归结为求解如下一个优化问题:
f(x)为势能函数,Ω为构象空间,有时为了简化计算,常固定某些自由度,将问题化为一个具有上下界约束的优化问题。由于蛋白质体系作用极其复杂,能量函数具有极多的局部极小点,所以如何找到势能函数的全局极小点是优化过程的一个关键。
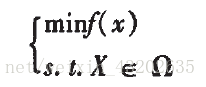
通常分两个步骤完成:
(1)产生初始点;
(2)采用典型的优化方法如最速下降法、共轭梯度法、牛顿法、拟牛顿法等进行能量极小化,重复这一过程直到求出满意解。
由于能量极小化过程只能找到初始点附近的局部极小点,初始点的产生对整个优化过程的效率起着重要作用。根据初始点产生的策略不同,常用的构象搜索算法可分为系统搜索法和随机搜索法。
2.1 系统搜索
即系统地搜索整个构象空间,寻找能量极小点。最基本的系统搜索方法是格点搜索,将整个构象空间划分成小间距网格,格点表示氨基酸的重心,逐个计算每一个格点对应的能量值。这种方法随着变量的增加,计算量急剧增加。其他方法还有过度算法、搭建过程、分支定界法等。
2.2 随机搜索
它可以在更大的构象空间寻找极小点,然而到目前为止,还没有一种有效的随机搜索法能保证在有效的计算时间内找到全局能量极小点。用于蛋白质构象搜索的随机算法主要有:Monte Carlo法、遗传算法、模拟退火算法、神经网络算法、禁忌搜索算法、图论算法等,关于这些方法的综述很多,本文不逐一介绍,这里主要讨论从头预测较成功的Monte Carlo法。
Monte Carlo法是一种常用的随机搜索方法,Martin Sander最早将该方法引入分子构象搜索后,它在蛋白质结构预测中得到了广泛应用,并发展了许多不同的策略。
它首先假设蛋白质的折叠过程是一个具有唯一吸收状态的Markov过程,利用Metropolis采样算法搜索构象。这个过程分为两个步骤:
(1)在高温下将体系加热熔化;
(2)逐渐降温。
在任何一个温度下,体系的初构象ri,相应锈量E(ri),构象发生微小的随机变化产生新的构象ri+1,相应的能量E(ri+1),能量变化为E=E(ri+1)一E(ri)。E不大于零时,接受构象变化,而当E大于零时,则被接受的几率为:P(△E)=e-△E/(kT),即在(0,1)之间选择一个随机数R,将其与P(△E)比较,若R<P(△E)则接受变化,新的构象成为下一次随机变化的起点,否则,拒绝变化,不断循环往复,便可以模拟出该温度下体系的波尔兹曼分布,最终达到系统内能的最小值状态,而不会在冷却过程中某一局部极小值点出现阻塞,模拟这一过程的优化模型称为
3. 模拟退火模型
该模型把优化过程与退火过程对应起来,建立起优化目标函数与系统内能、变量与分子结构的对应关系,它与局部优化下降算法最显著的区别是它能按一定概率接受使目标函数值增加的状态变化,因而它的动态特性能够使状态变量跳过局部优化点,最终达到全局最优点。这一状态变化的接受规则就是Monte Cado采用的Metropolis准则。
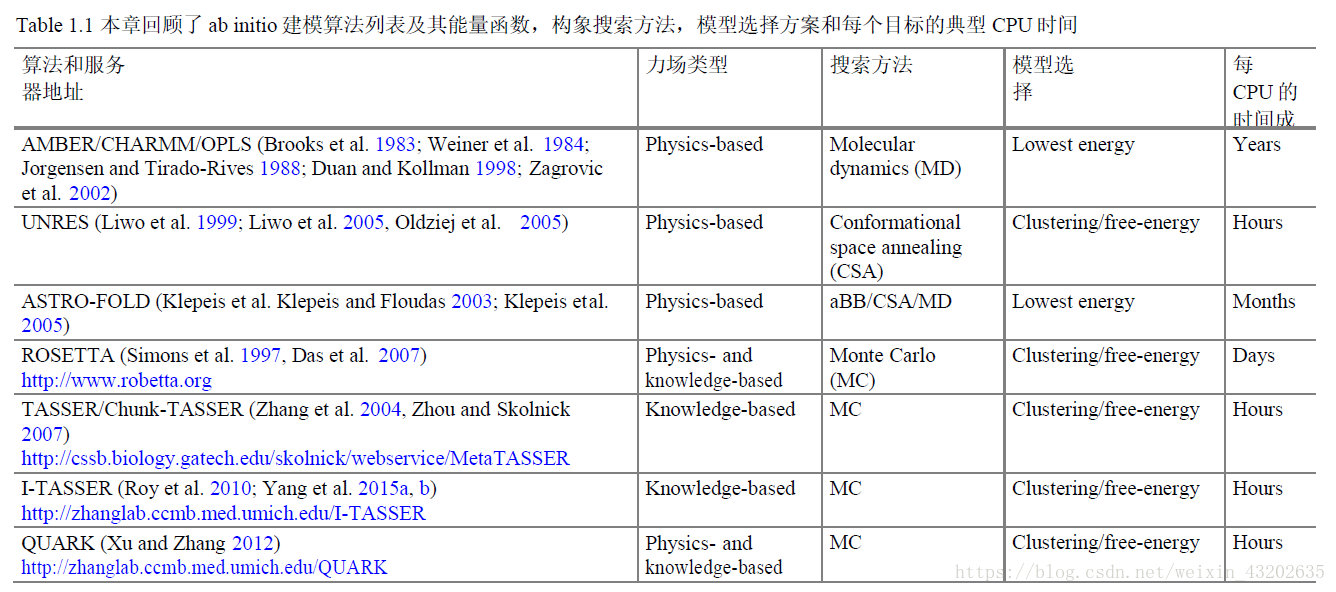
4. 成功的从头预测方法
碎片组装的方法是迄今最成功的从头预测方法,David Baker的ROSETTA就是基于这一理论建立的。它基于这样一个假设:短的序列片段局限在蛋白质结构数据库中最相关的序列局部结构。
分两个步骤:
(1) 将目标序列切割成互相重叠的小碎片,并产生每个碎片的局部二级结构;
(2) 用基于贝叶斯概率理论的能量函数和Monte Cdo片段插入法组装这些局部结构来产生模型,选择最小能量模型。
3.1 ROSETTA
David Baker及团队设计的ROSETTA方法是碎片组装理论的最佳体现,它把基于物理和统计学(贝叶斯概率论)知识能量函数结合,搜索策略是简化的Monte Carlo法。自CASP3上崭露头角以来,在以后的CASP中Rosetta的预测结果都不断接近真实值。
近年来的进展,除了修饰能量函数外主要体现在三方面:
(1)基础模拟方法:用许多不同的二级结构方法(PsIPRED,SAMT99,PHD)减少碎片选择偏差,利用一个简单的邻近表进行碎片取代,只计算改变的距离,减少了计算时间;碎片替代后产生的链之间的碰撞带来的波动,用局部运动或是改变骨架转角来解决;
(2)从模拟结构的大量集合中去除非蛋白质构象(差的beta片层、劣势堆积结构),提高近天然构象概率,对于分子量大的蛋白质,高接触序(higII contact order)结构从额外的模拟中选择;
(3)对一个给定目标序列的许多独立构象模拟聚类,通过识别最小能量值来多样化一个蛋白质家族,从某种程度上补偿能量函数的不精确性。
3.2 I-TASSER
与ROSElTTA齐头并进的还有在CASP5上张阳与Jeffrey Skolnick合作的TASSER,其采用基于知识的能量函数。与ROSETTA采取固定长度碎片(3或是9个残基)不同,虽然都是从PDB中选择碎片来组装,TASSER用不同长度片段通过穿线法产生结构模型,从中提取结构片段。在CASP7上,张阳单独设计了新的方法——I-TASSER,在TASSER基础上对碎片进行再切割和组装,大大提高了精确度。
3.3 Quark
QUARK的首要目的是在仅使用蛋白质序列信息的情况下实现蛋白质结构的准确预测。QUARK输出的模型使用长度为1-20片段的组装,并且使用基于知识的原子粒度力场模型引导的副本交换蒙塔卡罗模拟的搜索策略。
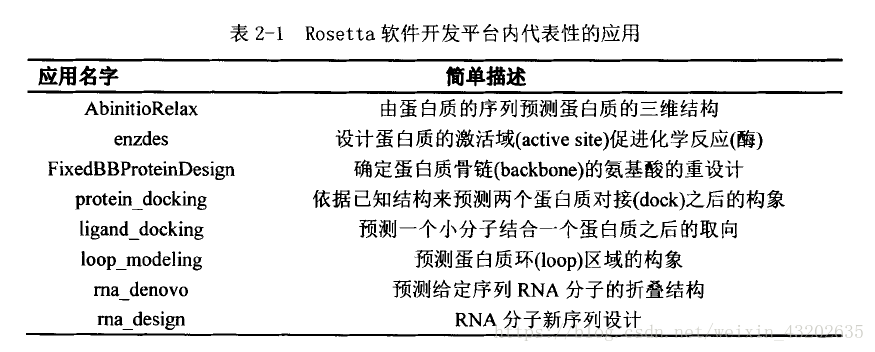
二.Rosetta策略
Robetta在线服务器是由Baker实验室提供的非商业使用的自动蛋白质结构预测服务器,其主要使用的方法是从头预测和比对建模,Robeaa使用Rosetta提供的软件包构建相应的服务。
在Rosetta算法中,通过局部序列采样的结构通过在已知蛋白质结构中看到的那些短序列和相关序列的结构分布来近似。链选自已知蛋白质结构的数据库。然后通过使用蒙特卡罗模拟退火搜索随机组合这些碎片来组装紧凑结构。基于来自已知蛋白质结构的构象统计学的评分函数评估个体构型相对于非局部相互作用的适合度。
注:片段库,其代表蛋白质的所有短片段的可接近的局部结构的范围
Rosetta设计目的是让用户定义特定的算法来解决特定的蛋白质分子建模问题。并且可以简单的通过使用PyRosetta库文件的Python脚本在Mac或者Linux平台来实现算法,概略流程正如图2.1所示。用户可以使用文献已经发表的,并在Rosetta中实现的标准算法的相关类结构以及功能辅助实现自身算法的设计。
PyRosetta是一个基于Python脚本语言实现Rosetta分子建模软件开发平台的接口设计。其大体的设计流程如图2.2所示。使用户更加容易的通过Python脚本来使用Rosetta的采样以及能量函数来创建自定义的分子建模算法。
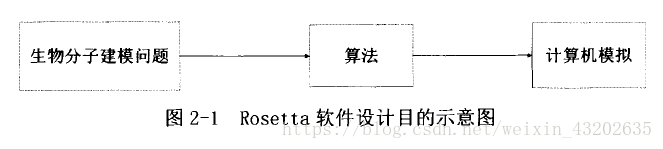
PyRosetta设计的实际目的是为了复杂的Rosetta软件开发平台让更多的人易于使用,尤其是生命科学领域以及其他领域不熟悉相应编程的研究人员。用户可以使用PyRosetta实现新的算法,也可以通过编辑使用Rosetta提供的类结构和方法的Python脚本来使用已有算法。本论文中实现的算法默认使用Rosetta的Python实现版本PyRosetta。
以残基为中心的Rosetta设计体现在两个方面:整个分子中的原子在残基之中体现出来,残基是能量函数计算的基本单位。

Rosetta是一个用于大分子建模的非常大的软件套件。通过软件套件,我们的意思是它是一个庞大的计算机代码集合(大部分是C ++,有些是Python,有些是其他语言)。
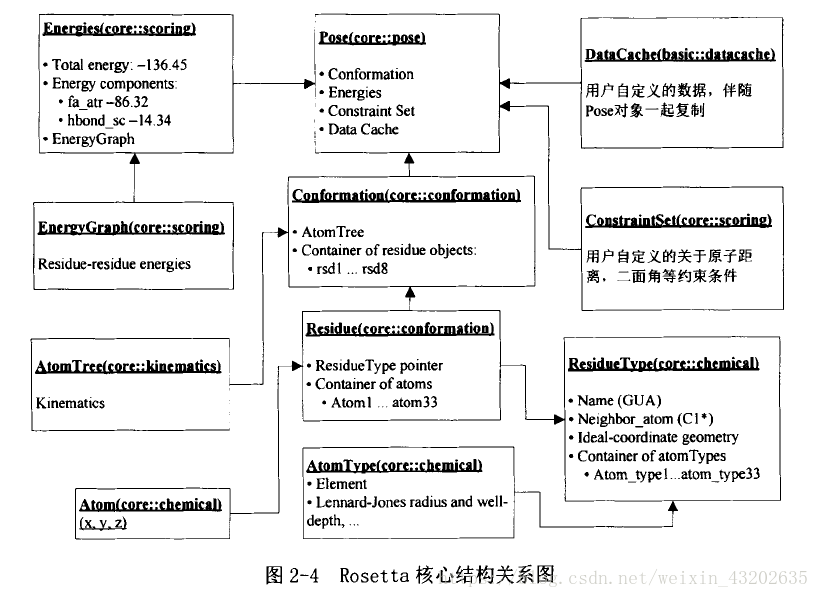
Rosetta软件的基本架构以Pose类作为核心类展开:
类Pose的对象在蛋白质结构预测方面代表一个蛋白质分子构象,在Pose类对象中包含其他相关类的引用,
蛋白质分子的构象空间信息和化学结构相关信息存储在Conformation类的包含Residue类对象的容器(Container)里面,
在Residue类里面包含指向ResidueType类的指针,来标明氨基酸残基的特定类型,
在AtomTree类对象记录了分子的运动学连接信息一一内部坐标系和笛卡尔坐标系之间的映射,
使用能量函数评价的最新能量值存储在Energies类对象内,在Energies内部包括了类EnergyGraph,这个类记录了残基.残基之间的交互能量,
用户定义的坐标约束条件存储在ConstraintSet类对象内,
对其他额外定义的Pose对象相关的数据可以存储在DataCache类对象内,存储在DataCache类对象内部的Pose类相关信息在模拟的整个过程中会随着Pose一起复制。














 590
590











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









