






打算仔细学习一下基于python的单细胞相关分析框架hhh
新手上路写的很繁琐,多多包涵,本次用的IDE是Visual studio code。
流程来自Scanpy官网(Preprocessing and clustering 3k PBMCs (legacy workflow)):
https://scanpy.readthedocs.io/en/stable/tutorials/basics/clustering-2017.html
步骤流程
1、check
# 当前工作目录
import os
# 当前工作目录
os.getcwd()
# '/Users/zaneflying/Desktop/scanpy'
在Python中,os 是一个标准库,提供了一系列的功能来与操作系统交互。这个包允许你执行例如文件和目录操作(如创建、删除、修改)、获取操作系统相关信息、处理文件路径等任务。它是Python的核心库之一,不需要额外安装,可以直接导入使用。这使得os包成为处理文件系统和操作系统任务时非常有用的工具
# 目录下文件
os.listdir()
# ['scanpy.ipynb', '.DS_Store', 'input', 'data']
2、导入
顺口一提,安装库的形式是这样的:pip install pandas
import pandas as pd
import scanpy as sc
import numpy as np
import anndata as ad
import pooch
Pandas 是一个开源的 Python 数据分析库,广泛用于快速分析数据,以及数据清洗和准备等工作。它提供了高效的 DataFrame 对象,使得在 Python 中可以方便地进行创建、操作和预处理结构化数据。Pandas 提供了大量易于使用的功能和方法,用于数据导入、转换、清理和可视化
3、分析前设置参数
sc.settings.verbosity = 3
sc.logging.print_header()
# scanpy==1.10.3 anndata==0.10.9 umap==0.5.6 numpy==2.0.2 scipy==1.13.1 pandas==2.2.3 scikit-learn==1.5.2 statsmodels==0.14.3 igraph==0.11.6 louvain==0.8.2 pynndescent==0.5.13
sc.settings.set_figure_params(dpi=80, facecolor="white")
上述代码学习:
sc.settings.verbosity?
# 在函数后面加上?再运行的时候就是查看函数的帮助文档
# 这个函数的作用拆解一下,sc是scanpy的简写,后面跟上的settings.verbosity 关于日志记录的详细等级,3表示显示提示
sc.logging.print_header?
# 此函数用于打印可能影响数值结果的各种依赖包的版本信息
sc.settings.set_figure_params?
# sc.settings.set_figure_params(
# *,
# scanpy: 'bool' = True,
# dpi: 'int' = 80,
# dpi_save: 'int' = 150,
# frameon: 'bool' = True,
# vector_friendly: 'bool' = True,
# fontsize: 'int' = 14,
# figsize: 'int | None' = None,
# color_map: 'str | None' = None,
# format: '_Format' = 'pdf',
# facecolor: 'str | None' = None,
# transparent: 'bool' = False,
# ipython_format: 'str' = 'png2x',
# ) -> 'None'
# 这个函数是设置图片的尺寸,这个跟R语言中就很不一样,一般R语言会在输出环节再设置。
# scanpy: 如果为True,使用Scanpy的默认matplotlib设置。
# dpi: 图像显示的分辨率。
# dpi_save: 保存图像的分辨率。
# frameon: 是否在图像周围显示框架。
# vector_friendly: 是否生成对矢量图友好的输出。
# fontsize: 图形中使用的字体大小。
# figsize: 图形的尺寸,如果为None,则使用默认尺寸。
# color_map: 使用的颜色映射。
# format: 保存图形时使用的格式。
# facecolor: 图形的背景颜色,如果为None,使用默认颜色。
# transparent: 是否使图形背景透明。
# ipython_format: 在Jupyter Notebook中显示图形时使用的格式。
3、设定一个文件输出路径
# 设定一个文件路径
results_file = "output/" # the file that will store the analysis results
4、读取文件
adata = sc.read_10x_mtx(
"input/", # the directory with the `.mtx` file
var_names="gene_symbols", # use gene symbols for the variable names (variables-axis index)
cache=True, # write a cache file for faster subsequent reading
)
到存放数据的文件夹,选择合适的读取方式,比如笔者这边使用的是10X的数据,所以选择sc.read_10x_mtx进行读取。
除此之外有很多其他的读取方式。
## Preprocessing
sc.read_10x_mtx?
# path (Path | str):
# 指定文件目录的路径,包含 .mtx 和 .tsv 文件。例如 './filtered_gene_bc_matrices/hg19/'。
# var_names (Literal['gene_symbols', 'gene_ids'], optional, default: 'gene_symbols'):
# 定义变量索引的方式,可以选择 'gene_symbols' 或 'gene_ids'。默认是 'gene_symbols'。
# make_unique (bool, optional, default: True):
# 是否确保变量索引唯一(通过在重复项后面加上 '-1', '-2' 等)。默认值是 True。
# cache (bool, optional, default: False):
# 是否缓存读取的文件。默认值是 False。
# cache_compression (Literal['gzip', 'lzf'] | None | Empty, optional):
# 指定缓存压缩的格式(如 'gzip' 或 'lzf'),默认值为 _empty,即没有指定。
# gex_only (bool, optional, default: True):
# 如果为 True,只会读取基因表达(gene expression, GEX)数据。默认值是 True。
# prefix (str | None, optional):
# 可以在文件名前添加前缀,默认为 None。
# 函数返回值
# 返回值 (AnnData):
# 函数返回一个 AnnData 对象,这是一个用于存储多维数组数据的常见结构,通常用于单细胞 RNA-seq 数据分析。
check一下adata
adata
AnnData object with n_obs × n_vars = 8931 × 33538:
n_obs: 代表观测的数量,即单细胞 RNA-seq 数据中的细胞数。在这个例子中,有 8931 个细胞(n_obs = 8931)。
n_vars: 代表变量的数量,即测量的基因数。在这个例子中,有 33538 个基因(n_vars = 33538)。
结合起来,表示这是一个 8931 个细胞 × 33538 个基因 的表达矩阵。
var: 'gene_ids', 'feature_types':
var 是 AnnData 对象中与变量(基因)相关的注释信息,这里列出了两个注释字段:
'gene_ids': 每个基因对应的基因 ID。
'feature_types': 基因的特征类型(如蛋白编码基因、非编码 RNA 等)。
这些注释字段存储了关于每个基因的元数据信息,可以在分析过程中用来进行筛选、分组等操作。
5、使变成唯一基因名
adata.var_names_make_unique()
如果是gene_ids就不需要做这一步,但是建议大伙们都加上。笔者这边使用的是gene_symbols
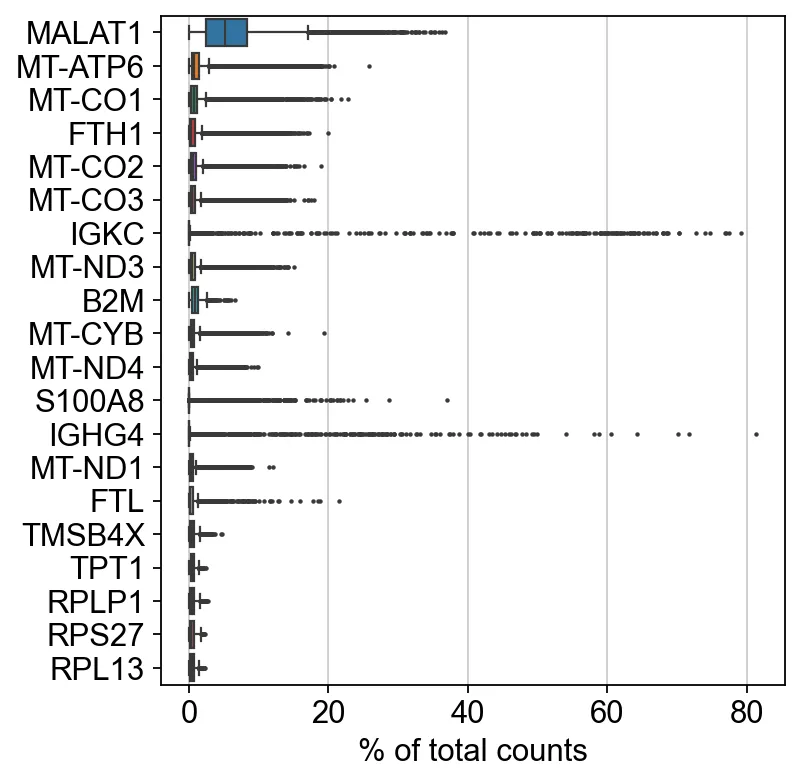
6、找一下前20的高表达基因
sc.pl.highest_expr_genes(adata, n_top=20)
sc代表scanpy,pl代表plot。这句代码在很多场合都可以用到
7、过滤数据
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# filtered out 196 cells that have less than 200 genes expressed
# filtered out 11773 genes that are detected in less than 3 cells
8、增加一列线粒体基因
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
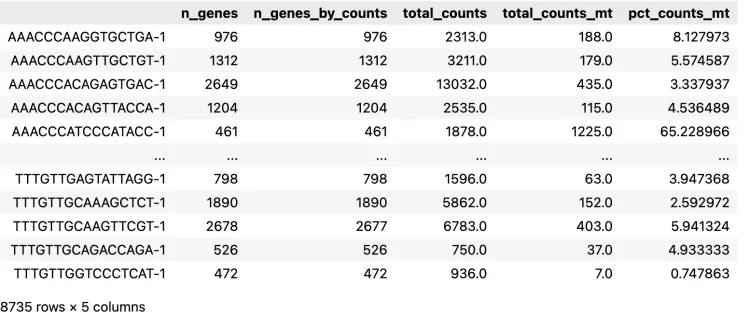
adata.obs
check一下观测数据
adata.var_names.str.startswith?
# adata 是一个 AnnData 对象,它是用于存储和操作单细胞 RNA 测序数据的主要数据结构。
# var_names 是 adata 对象的一个属性,表示数据集中基因的名称或 ID(即变量名称)。具体来说,它是一个 pandas.Index 对象,包含所有基因的名称或 ID。
# .str 是 pandas 提供的一个字符串操作方法的访问器,用于对 pandas.Series 或 Index 中的字符串进行矢量化操作。
# 通过 .str,您可以对 var_names 中的所有基因名称(字符串)同时进行各种字符串操作,例如检查前缀、后缀、包含关系、替换子字符串等。
# .startswith 是一个字符串方法,用于检查字符串是否以指定的前缀开头。
# 当与 .str 结合使用时,startswith 可以用来检查 var_names 中的每一个基因名称是否以某个特定的前缀开头。
sc.pp.calculate_qc_metrics?
# qc_vars 参数指定要用于质量控制的基因类别或变量。通常这是一个列表,包含用于识别特定基因集的前缀或关键字。
# 在这里,["mt"] 表示线粒体基因(通常以 "MT-" 或类似的前缀开头的基因)。这意味着该函数将计算与线粒体基因相关的 QC 指标。
# 这个参数指定要计算的前 n 个表达最多的基因占总表达量的百分比。例如,percent_top=[50, 100] 会计算表达最多的前 50 个和前 100 个基因的总表达百分比。
# 在这里设置为 None,意味着不计算这个指标。
# log1p 参数决定是否在计算 QC 指标之前对数据进行对数转换 (log(1 + x))。
# 在这里,log1p=False 表示不对数据进行对数转换。
# 这个参数决定函数是否直接在输入的 adata 对象中添加计算出的 QC 指标。
# inplace=True 表示计算出的 QC 指标将直接添加到 adata 对象中,而不是返回一个新的对象。
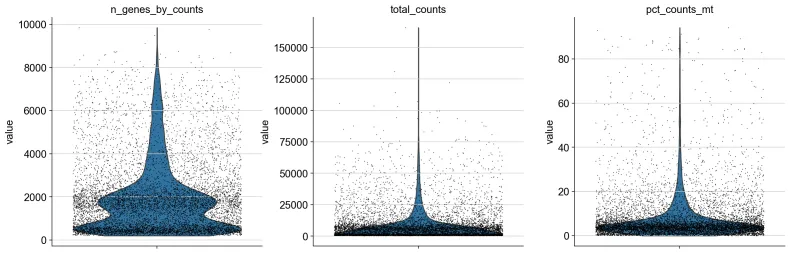
9、小提琴图绘制
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
sc.pl.violin?
# ["n_genes_by_counts", "total_counts", "pct_counts_mt"]:
# 这是一个字符串列表,指定要绘制的小提琴图的变量名称:
# n_genes_by_counts: 每个细胞检测到的基因数量(通常指非零表达的基因数量)。这是衡量细胞复杂度的一个指标。
# total_counts: 每个细胞的总 UMI(Unique Molecular Identifier)计数,表示每个细胞中的总 RNA 量。
# pct_counts_mt: 线粒体基因的表达量占总表达量的百分比,通常用于检测死细胞或低质量细胞。
# 这些变量通常是在质量控制(QC)步骤中生成的。
# jitter=0.4:
# jitter 参数控制散点图中的点在 x 轴上的抖动量。抖动可以防止点重叠,使数据分布更清晰可见。
# 在这里,jitter=0.4 表示将点轻微地分散,以避免重叠。
# multi_panel=True:
# multi_panel 参数决定是否将多个变量绘制在不同的小提琴图面板中。
# multi_panel=True 表示将每个变量分别绘制在不同的小提琴图面板中,而不是将所有变量显示在同一个图中。
10、scatter图绘制
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")

11、进一步过滤
把count数在2500和线粒体百分比在5以上的信息全过滤掉。
使用.copy从解释来看是不会影响这一步之前的数据(这里笔者还没有一个清晰认识,但是意思是这个意思)
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
adata = adata[adata.obs.n_genes_by_counts < 2500, :]:
# adata.obs.n_genes_by_counts:这是一个包含每个细胞中检测到的基因数量的列,存储在 AnnData 对象的 obs 数据框中。
# adata[adata.obs.n_genes_by_counts < 2500, :]:这行代码的作用是过滤出那些检测到的基因数量小于 2500 的细胞。
# adata.obs.n_genes_by_counts < 2500 生成一个布尔向量,标识哪些细胞的基因数量少于 2500。
# adata[...] 使用这个布尔向量来选择符合条件的细胞,将不符合条件的细胞移除。
# 结果:保留基因数量少于 2500 的细胞,过滤掉其他细胞。
# adata = adata[adata.obs.pct_counts_mt < 5, :].copy():
# adata.obs.pct_counts_mt:这是一个包含每个细胞中线粒体基因表达占总表达比例的列。
# adata[adata.obs.pct_counts_mt < 5, :]:这行代码进一步筛选出线粒体基因表达比例小于 5% 的细胞。
# adata.obs.pct_counts_mt < 5 生成一个布尔向量,标识哪些细胞的线粒体基因表达比例小于 5%。
# adata[...] 使用这个布尔向量来选择符合条件的细胞,将不符合条件的细胞移除。
# .copy():这个方法创建了一个 adata 对象的副本,确保后续操作不会影响原始数据。
12、Normalize数据
这句代码的参数就跟seruat中很像了
sc.pp.normalize_total(adata, target_sum=1e4)
13、Normalize之后还需要对数据取log处理
log1p 是 log(1 + x) 的缩写,表示对每个数值 x 先加 1,然后取自然对数。
sc.pp.log1p(adata)
Seruat中是把上述两步整合在一起了。
14、找高变基因
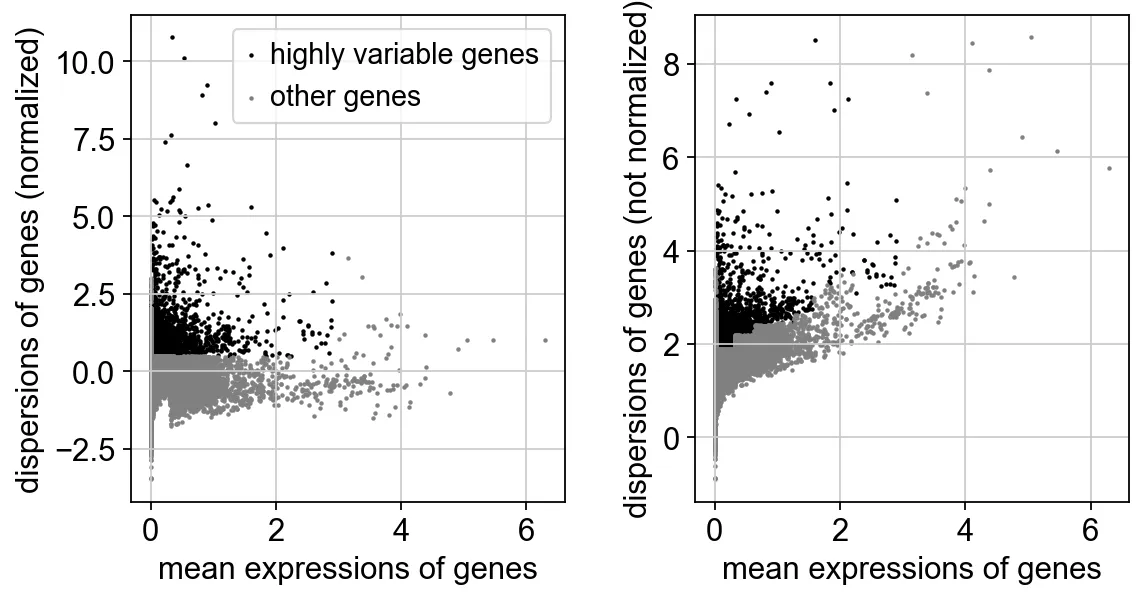
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# min_mean 参数设置了基因被认为是高度可变的最小平均表达阈值。也就是说,只有那些平均表达值超过 0.0125 的基因才会被考虑为候选的高度可变基因。
# max_mean 参数设置了基因被认为是高度可变的最大平均表达阈值。平均表达值高于 3 的基因将被排除在外。
# 这通常是为了避免选择那些过度表达的基因,这些基因可能会主导分析结果,但并不代表生物学上有意义的变化。
# min_disp 参数设置了基因变异度(dispersion)的最小阈值。只有那些变异度超过 0.5 的基因才会被考虑为候选的高度可变基因。
变异度是指基因表达值相对于其均值的标准化方差,变异度较高的基因在不同的细胞群体中表达差异较大。
sc.pl.highly_variable_genes(adata)
# sc 是 scanpy 的简称,代表整个库。
# pl 是 plot 的缩写,表示这是一个用于绘图的模块或函数。
# scanpy 库中按照功能模块进行了分层设计,其中:
# sc.pp (pp 是 preprocessing 的缩写):用于数据的预处理步骤,例如归一化、数据过滤、识别高度可变基因等。
# sc.tl (tl 是 tools 的缩写):用于各种分析工具的调用,例如 PCA、聚类、UMAP、差异表达分析等。
# sc.pl (pl 是 plot 的缩写):用于数据的可视化,例如绘制 PCA 图、UMAP 图、热图、小提琴图等。
绘制高可变基因的散点图
15、Scale数据
sc.pp.scale(adata, max_value=10)
# sc.pp.scale(adata, max_value=10) 对 adata 对象中的基因表达数据进行标准化,并将标准化后的数据限制在 [-10, 10] 的范围内
16、PCA分析,降维
sc.tl.pca(adata, svd_solver="arpack")
画一下PCA图
sc.pl.pca(adata, color="CST3")

17、显示每个主成分对数据的贡献
sc.pl.pca_variance_ratio(adata, log=True)

18、计算邻域图
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
umap
sc.tl.umap(adata, init_pos='paga')
sc.pl.umap(adata, color=["CST3", "NKG7", "PPBP"])
根据这些基因绘制umap图片

19、聚类
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
# random_state 参数用于设置随机种子,以确保聚类结果的可重复性。设置为 0 表示在相同数据和设置下,多次运行该算法将产生相同的结果。这是为了在多次运行时保持结果一致性,尤其是在需要重复实验或共享结果时。
# flavor 参数指定了使用哪种实现方式。"igraph" 是一种基于 igraph 库的实现,这是 Leiden 算法的默认实现。igraph 是一个高效的图处理库,通常用于图和网络分析。它支持快速的社区检测(如 Leiden 聚类)。
# n_iterations 参数设置算法的最大迭代次数。每次迭代都会尝试优化图的模块性,从而改进社区划分。n_iterations=2 表示最多进行 2 次迭代,通常用于平衡计算效率和聚类结果的质量。如果算法在早期迭代中已经收敛,可能不需要达到最大迭代次数。
# directed 参数指定图是否为有向图。如果设置为 False,则视为无向图。在大多数单细胞 RNA-seq 数据分析中,细胞相似性图通常被视为无向图,因此该参数通常设置为 False
sc.pl.umap(adata, color=["leiden"])
resolution=0.9时的聚类图,可以分成17个簇

整理一下代码
# 加载库
import pandas as pd
import scanpy as sc
import numpy as np
import anndata as ad
import pooch
# 参数设置
sc.settings.verbosity = 3
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor="white")
# 设定一个文件路径
results_file = "output/"
# 读取数据
adata = sc.read_10x_mtx(
"input/",
var_names="gene_symbols",
cache=True,
)
# 让基因名称不重复
adata.var_names_make_unique()
# 基础过滤
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# 增加线粒体基因数据
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
# 基础数据绘图
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4,
multi_panel=True,
)
# 基础数据绘图
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt")
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts")
# 过滤
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
# 根据上述信息可以调整过滤参数
##################################
# Normilize+log数据
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 寻找高变基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# 高变基因绘图
sc.pl.highly_variable_genes(adata)
# 备份一下
adata.raw = adata
# scale数据
sc.pp.scale(adata, max_value=10)
# pca降维
sc.tl.pca(adata, svd_solver="arpack")
# 计算邻域图+umap处理
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
sc.tl.umap(adata)
#################
# leiden聚类
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -















 1653
1653

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









