







先自我介绍一下,小编浙江大学毕业,去过华为、字节跳动等大厂,目前在阿里
深知大多数程序员,想要提升技能,往往是自己摸索成长,但自己不成体系的自学效果低效又漫长,而且极易碰到天花板技术停滞不前!
因此收集整理了一份《2024年最新Linux运维全套学习资料》,初衷也很简单,就是希望能够帮助到想自学提升又不知道该从何学起的朋友。

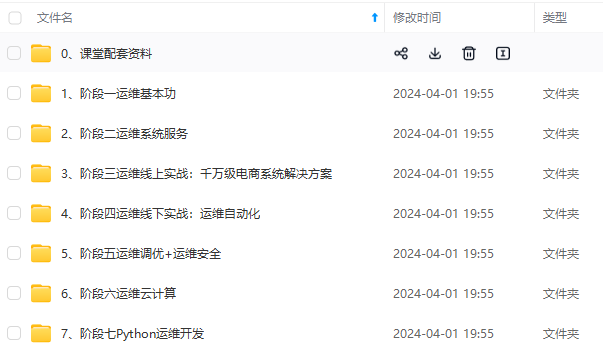
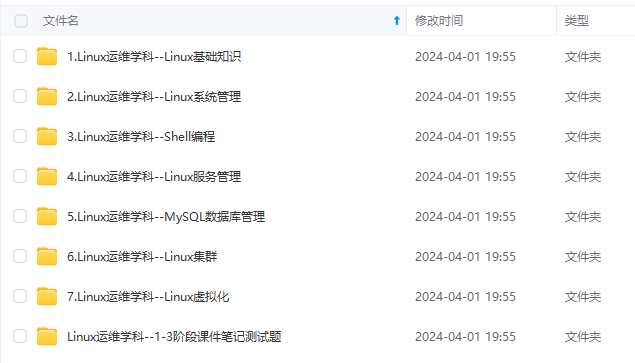
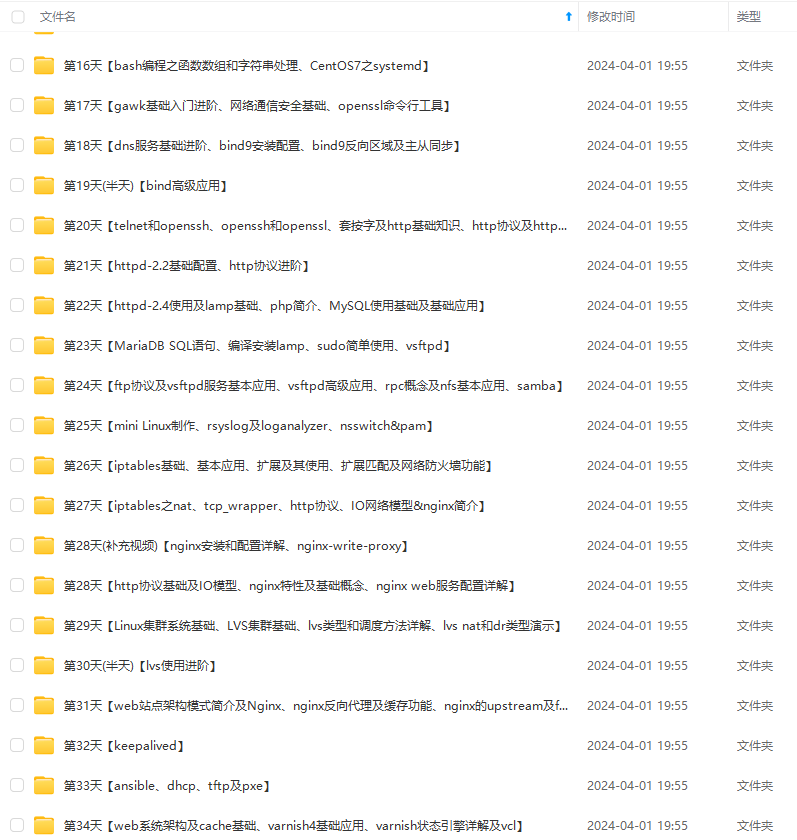
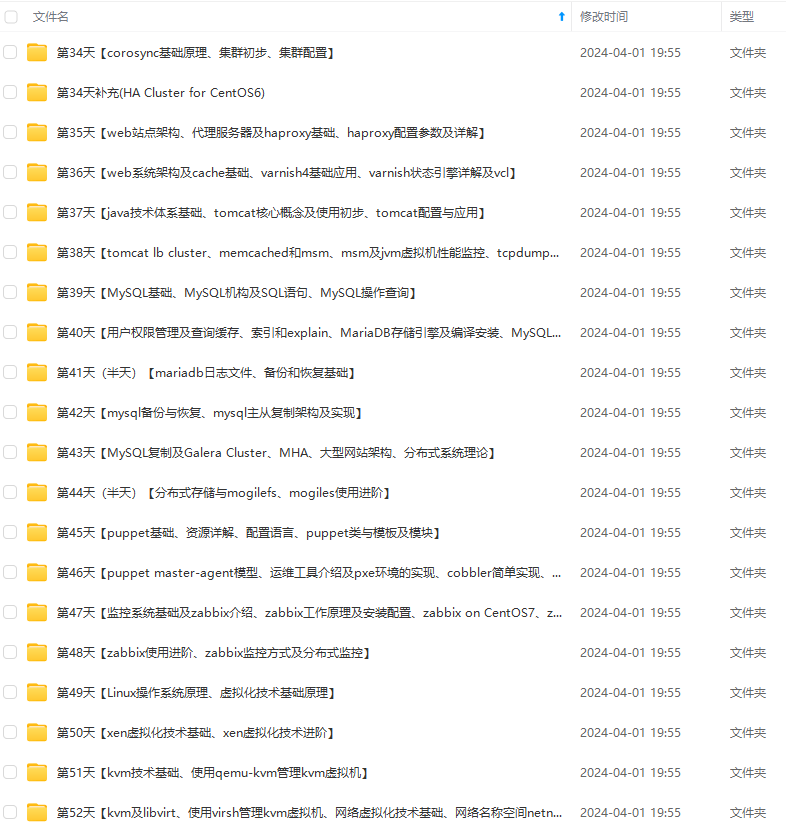
既有适合小白学习的零基础资料,也有适合3年以上经验的小伙伴深入学习提升的进阶课程,涵盖了95%以上运维知识点,真正体系化!
由于文件比较多,这里只是将部分目录截图出来,全套包含大厂面经、学习笔记、源码讲义、实战项目、大纲路线、讲解视频,并且后续会持续更新
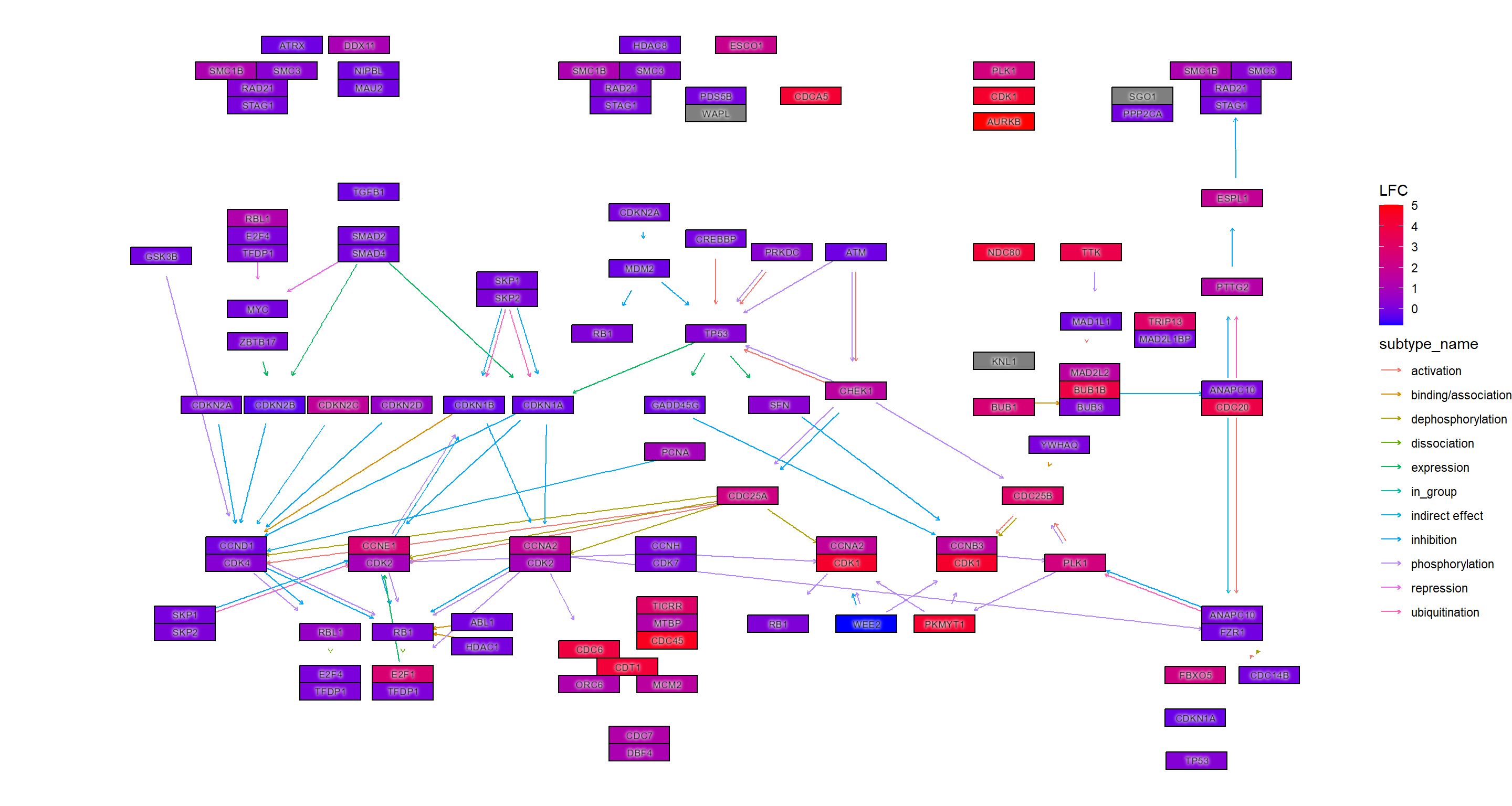
## Adjusted p-values
ggraph(g, layout="manual", x=x, y=y) +
geom_edge_parallel(width=0.5, arrow = arrow(length = unit(1, 'mm')),
start_cap = square(1, 'cm'),
end_cap = square(1.5, 'cm'), aes(color=subtype_name))+
geom_node_rect(aes(fill=padj, filter=type=="gene"), color="black")+
ggfx::with_outer_glow(geom_node_text(aes(label=converted_name, filter=type!="group"), size=2.5), colour="white", expand=1)+
scale_fill_gradient(name="padj")+
theme_void()
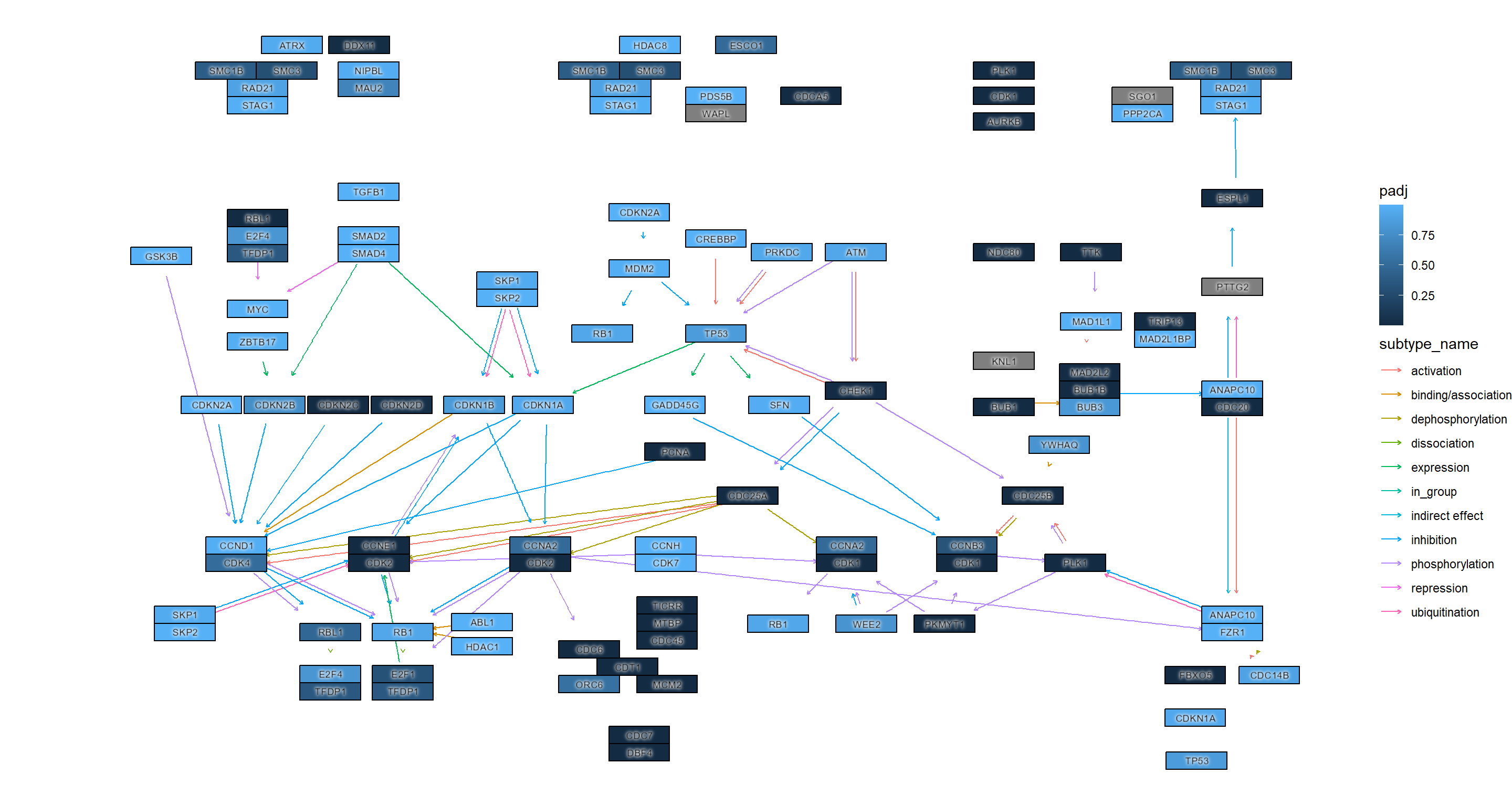
用于进一步自定义可视化ggfx
## Highlighting differentially expressed genes at adjusted p-values < 0.05 with coloring of adjusted p-values on raw KEGG map
gg <- ggraph(g, layout="manual", x=x, y=y)+
geom_node_rect(aes(fill=padj, filter=type=="gene"))+
ggfx::with_outer_glow(geom_node_rect(aes(fill=padj, filter=!is.na(padj) & padj<0.05)),
colour="yellow", expand=2)+
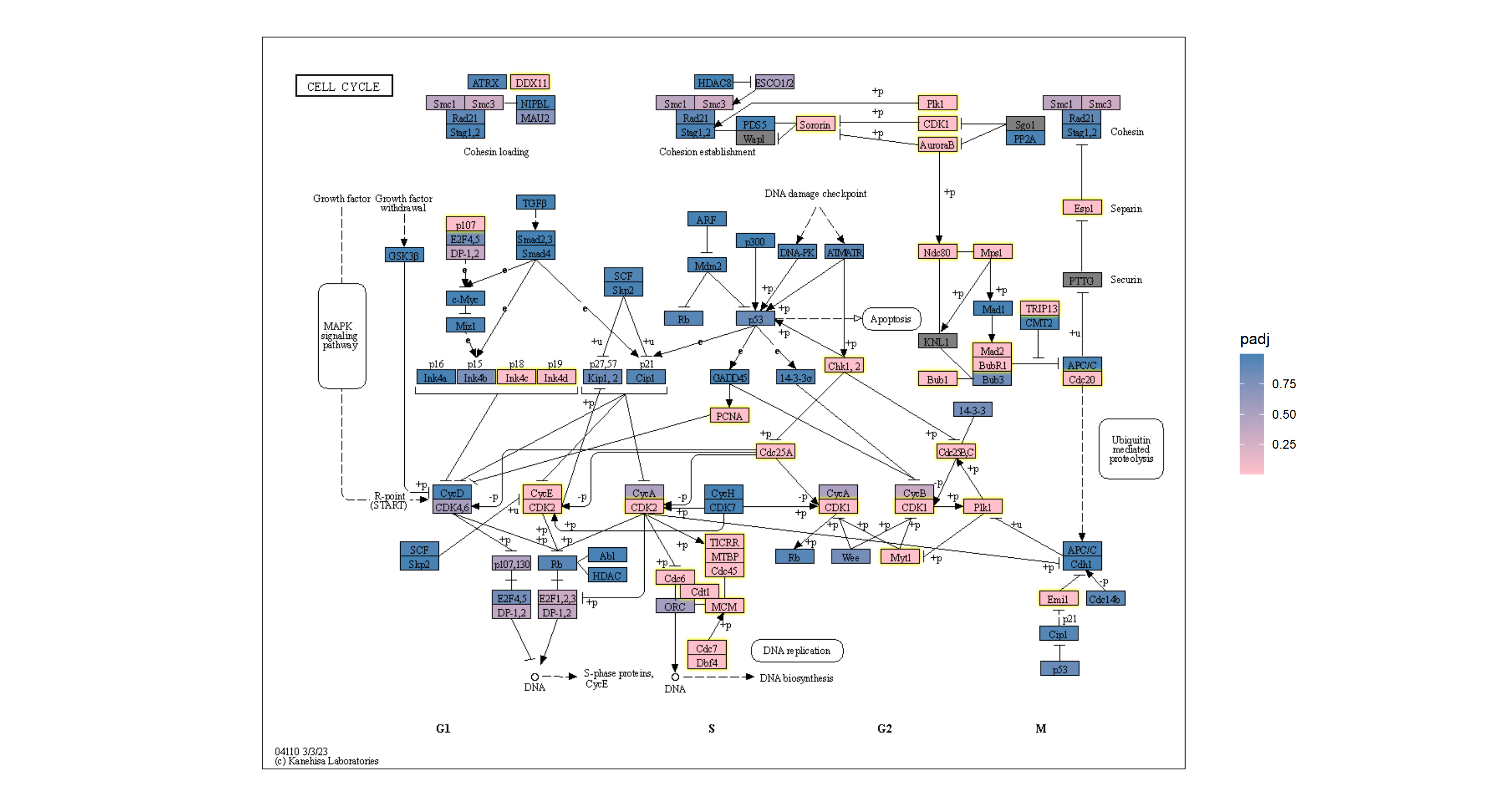
overlay_raw_map("hsa04110", transparent_colors = c("#cccccc","#FFFFFF","#BFBFFF","#BFFFBF"))+
scale_fill_gradient(low="pink",high="steelblue") + theme_void()
gg
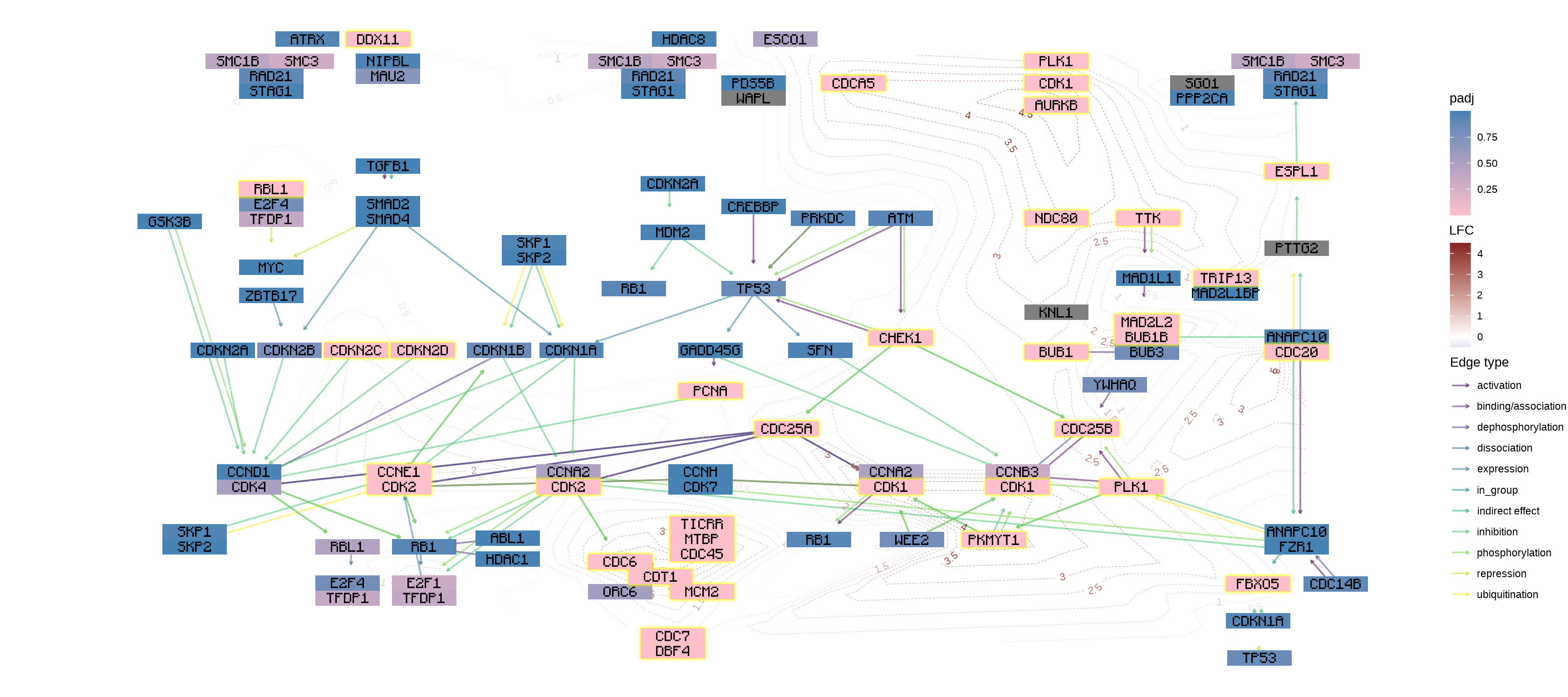
使用多个几何添加信息
您可以使用自己喜欢的几何图形及其扩展来添加信息。在此示例中,我们使用 geomtextpath 将 log2 折叠更改添加为轮廓,并使用 Monocraft 自定义字体。ggplot2
g <- g |> mutate(lfc=assign_deseq2(vinf, column="log2FoldChange"))
## Make contour data
df <- g |> data.frame()
df <- df[!is.na(df$lfc),]
cont <- akima::interp2xyz(interp::interp(df$x, df$y, df$lfc)) |>
data.frame() |> `colnames<-`(c("x","y","z"))
##
sysfonts::font_add(family="monocraft",regular="Monocraft.ttf")
gg <- ggraph(g, layout="manual", x=x, y=y)+
geom_edge_parallel(arrow=arrow(length=unit(1,"mm")),
aes(color=subtype_name),
end_cap=circle(7.5,"mm"),
alpha=0.5)+
geomtextpath::geom_textcontour(aes(x=x, y=y, z=z,color=after_stat(level)),
size=3, linetype=2,
linewidth=0.1, data=cont)+
geom_node_rect(aes(fill=padj, filter=type=="gene"))+
ggfx::with_outer_glow(geom_node_rect(aes(fill=padj, filter=!is.na(padj) & padj<0.05)),
colour="yellow", expand=2)+
geom_node_text(aes(label=converted_name), family="monocraft")+
scale_color_gradient2(low=scales::muted("blue"),
high=scales::muted("red"),
name="LFC")+
scale_edge_color_manual(values=viridis::viridis(11), name="Edge type")+
scale_fill_gradient(low="pink",high="steelblue") +
theme_void()
gg
将数值积分到tbl_graph
将数值向量积分到tbl_graph
数值可以反映在节点或边表中,利用 或 函数。输入可以是命名向量,也可以是包含 id 和 value 列的 tibble。node_numeric``edge_numeric
vec <- 1
names(vec) <- c("hsa:51343")
new_g <- g |> mutate(num=node_numeric(vec))
new_g
#> # A tbl_graph: 134 nodes and 157 edges
#> #
#> # A directed acyclic multigraph with 40 components
#> #
#> # A tibble: 134 × 23
#> name type reaction graphics_name x y width
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 hsa:1029 gene <NA> CDKN2A, ARF,… 532 -218 46
#> 2 hsa:51343 gene <NA> FZR1, CDC20C… 981 -630 46
#> 3 hsa:4171 h… gene <NA> MCM2, BM28, … 553 -681 46
#> 4 hsa:23594 … gene <NA> ORC6, ORC6L.… 494 -681 46
#> 5 hsa:10393 … gene <NA> ANAPC10, APC… 981 -392 46
#> 6 hsa:10393 … gene <NA> ANAPC10, APC… 981 -613 46
#> # ℹ 128 more rows
#> # ℹ 16 more variables: height <dbl>, fgcolor <chr>,
#> # bgcolor <chr>, graphics_type <chr>, coords <chr>,
#> # xmin <dbl>, xmax <dbl>, ymin <dbl>, ymax <dbl>,
#> # orig.id <chr>, pathway_id <chr>, deseq2 <dbl>,
#> # padj <dbl>, converted_name <chr>, lfc <dbl>, num <dbl>
#> #
#> # A tibble: 157 × 6
#> from to type subtype_name subtype_value pathway_id
#> <int> <int> <chr> <chr> <chr> <chr>
#> 1 118 39 GErel expression --> hsa04110
#> 2 50 61 PPrel inhibition --| hsa04110
#> 3 50 61 PPrel phosphorylation +p hsa04110
#> # ℹ 154 more rows
将矩阵积分到tbl_graph
如果要在图形中反映表达式矩阵,则 和 函数可能很有用。通过指定基质和基因 ID,您可以将每个样品的数值分配给 . 分配由边连接的两个节点的总和,忽略组节点(Adnan 等人,2020 年)。edge_matrix``node_matrix``tbl_graph``edge_matrix
mat <- assay(vst(res))
new_g <- g |> edge_matrix(mat) |> node_matrix(mat)
new_g
#> # A tbl_graph: 134 nodes and 157 edges
#> #
#> # A directed acyclic multigraph with 40 components
#> #
#> # A tibble: 134 × 48
#> name type reaction graphics_name x y width
#> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 hsa:1029 gene <NA> CDKN2A, ARF,… 532 -218 46
#> 2 hsa:51343 gene <NA> FZR1, CDC20C… 981 -630 46
#> 3 hsa:4171 h… gene <NA> MCM2, BM28, … 553 -681 46
#> 4 hsa:23594 … gene <NA> ORC6, ORC6L.… 494 -681 46
#> 5 hsa:10393 … gene <NA> ANAPC10, APC… 981 -392 46
#> 6 hsa:10393 … gene <NA> ANAPC10, APC… 981 -613 46
#> # ℹ 128 more rows
#> # ℹ 41 more variables: height <dbl>, fgcolor <chr>,
#> # bgcolor <chr>, graphics_type <chr>, coords <chr>,
#> # xmin <dbl>, xmax <dbl>, ymin <dbl>, ymax <dbl>,
#> # orig.id <chr>, pathway_id <chr>, deseq2 <dbl>,
#> # padj <dbl>, converted_name <chr>, lfc <dbl>,
#> # SRR14509882 <dbl>, SRR14509883 <dbl>, …
#> #
#> # A tibble: 157 × 34
#> from to type subtype_name subtype_value pathway_id
#> <int> <int> <chr> <chr> <chr> <chr>
#> 1 118 39 GErel expression --> hsa04110
#> 2 50 61 PPrel inhibition --| hsa04110
#> 3 50 61 PPrel phosphorylation +p hsa04110
#> # ℹ 154 more rows
#> # ℹ 28 more variables: from_nd <chr>, to_nd <chr>,
#> # SRR14509882 <dbl>, SRR14509883 <dbl>,
#> # SRR14509884 <dbl>, SRR14509885 <dbl>,
#> # SRR14509886 <dbl>, SRR14509887 <dbl>,
#> # SRR14509888 <dbl>, SRR14509889 <dbl>,
#> # SRR14509890 <dbl>, SRR14509891 <dbl>, …
边值
相同的效果可以通过 获得,使用命名数值向量作为输入。此函数根据节点值添加边值。以下示例显示了将 LFC 组合到边缘。这与 的行为不同。edge_matrix``edge_numeric_sum``edge_numeric
## Numeric vector (name is SYMBOL)
vinflfc <- vinf$log2FoldChange |> setNames(row.names(vinf))
g |>
## Use graphics_name to merge
mutate(grname=strsplit(graphics_name, ",") |> vapply("[", 1, FUN.VALUE="a")) |>
activate(edges) |>
mutate(summed = edge_numeric_sum(vinflfc, name="grname")) |>
filter(!is.na(summed)) |>
activate(nodes) |>
mutate(x=NULL, y=NULL, deg=centrality_degree(mode="all")) |>
filter(deg>0) |>
ggraph(layout="nicely")+
geom_edge_parallel(aes(color=summed, width=summed,
linetype=subtype_name),
arrow=arrow(length=unit(1,"mm")),
start_cap=circle(2,"mm"),
end_cap=circle(2,"mm"))+
geom_node_point(aes(fill=I(bgcolor)))+
geom_node_text(aes(label=grname,
filter=type=="gene"),
repel=TRUE, bg.colour="white")+
scale_edge_width(range=c(0.1,2))+
scale_edge_color_gradient(low="blue", high="red", name="Edge")+
theme_void()

可视化多重富集结果
您可以可视化多个富集分析的结果。与将函数与类一起使用类似,可以在函数中使用一个函数。通过向此功能提供对象,如果结果中存在可视化的通路,则通路内的基因信息可以反映在图中。在这个例子中,除了上面提到的尿路上皮细胞的变化外,还比较了肾近端肾小管上皮细胞的变化(Assetta等人,2016)。ggkegg``enrichResult``append_cp``mutate``enrichResult
## These are RDAs storing DEGs
load("degListRPTEC.rda")
load("degURO.rda")
library(org.Hs.eg.db);
library(clusterProfiler);
input_uro <- bitr(uroUp, ## DEGs in urothelial cells
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)$ENTREZID
input_rptec <- bitr(gls$day3_up_rptec, ## DEGs at 3 days post infection in RPTECs
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)$ENTREZID
ekuro <- enrichKEGG(gene = input_uro)
ekrptec <- enrichKEGG(gene = input_rptec)
g1 <- pathway("hsa04110") |> mutate(uro=append_cp(ekuro, how="all"),
rptec=append_cp(ekrptec, how="all"),
converted_name=convert_id("hsa"))
ggraph(g1, layout="manual", x=x, y=y) +
geom_edge_parallel(width=0.5, arrow = arrow(length = unit(1, 'mm')),
start_cap = square(1, 'cm'),
end_cap = square(1.5, 'cm'), aes(color=subtype_name))+
geom_node_rect(aes(fill=uro, xmax=x, filter=type=="gene"))+
geom_node_rect(aes(fill=rptec, xmin=x, filter=type=="gene"))+
scale_fill_manual(values=c("steelblue","tomato"), name="urothelial|rptec")+
ggfx::with_outer_glow(geom_node_text(aes(label=converted_name, filter=type!="group"), size=2), colour="white", expand=1)+
theme_void()
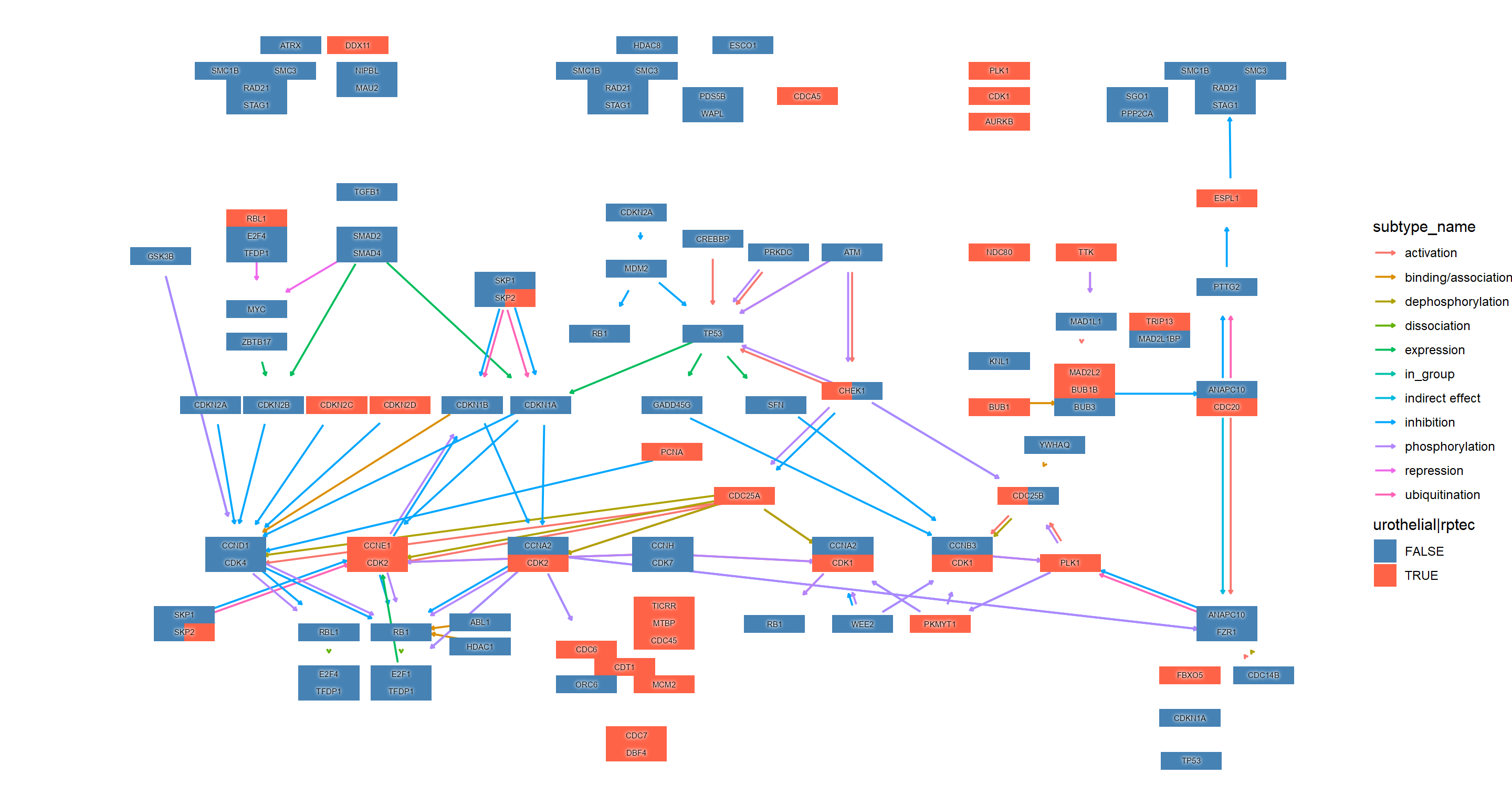
我们可以按 组合多个图。rawMap``patchwork
library(patchwork)
comb <- rawMap(list(ekuro, ekrptec), fill_color=c("tomato","tomato"), pid="hsa04110") +
rawMap(list(ekuro, ekrptec), fill_color=c("tomato","tomato"),
pid="hsa03460")
comb
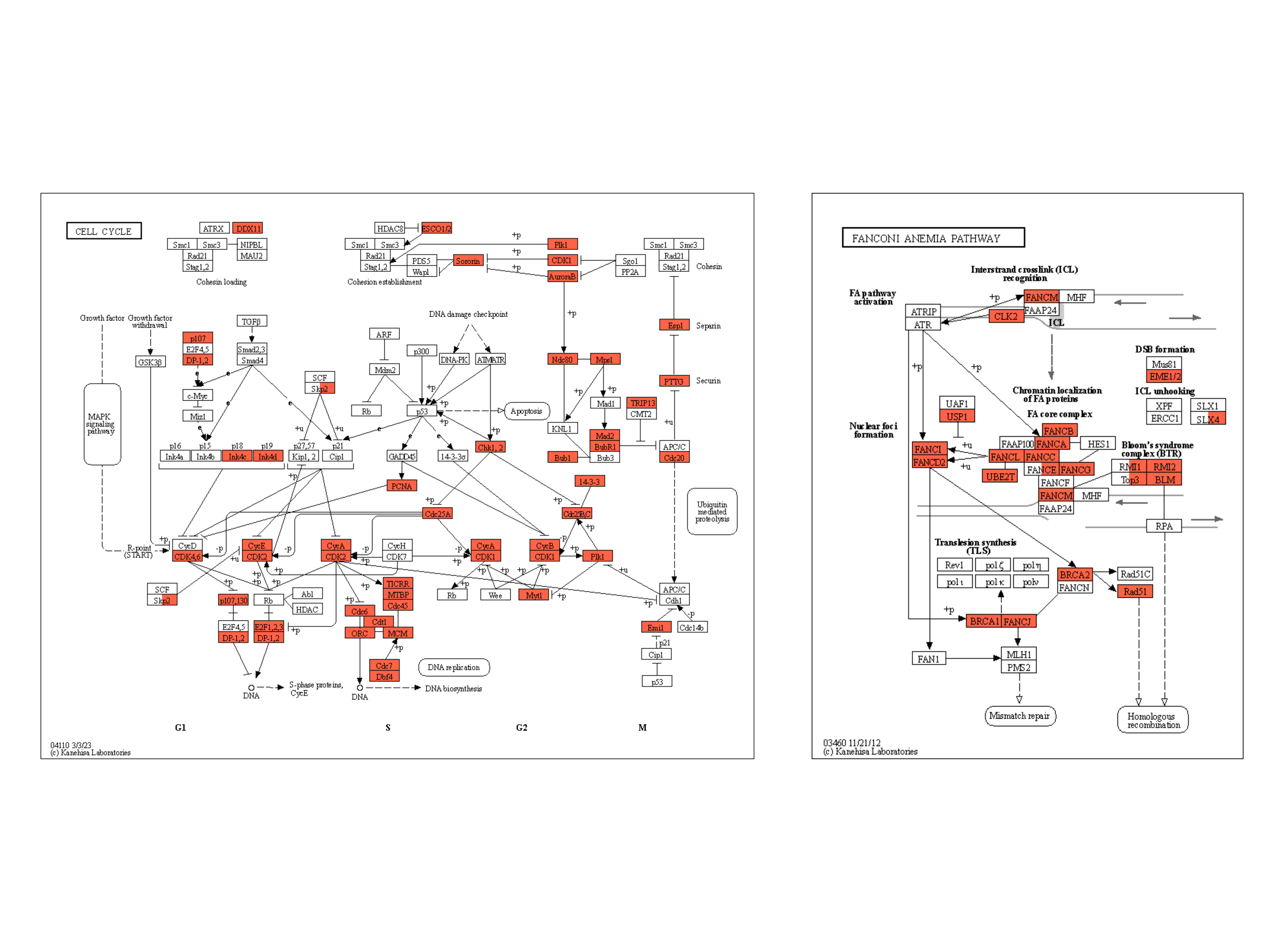
下面的示例将类似的反射应用于原始 KEGG 图谱,并突出显示在两种条件下都显示出统计学显着变化的基因,使用黄色外光,由 clusterProfiler 生成的组成,富集结果为 。ggfx``dotplot``patchwork
right <- (dotplot(ekuro) + ggtitle("Urothelial")) /
(dotplot(ekrptec) + ggtitle("RPTECs"))
g1 <- pathway("hsa03410") |>
mutate(uro=append_cp(ekuro, how="all"),
rptec=append_cp(ekrptec, how="all"),
converted_name=convert_id("hsa"))
gg <- ggraph(g1, layout="manual", x=x, y=y)+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=uro&rptec),
color="gold", fill="transparent"),
colour="gold", expand=5, sigma=10)+
geom_node_rect(aes(fill=uro, filter=type=="gene"))+
geom_node_rect(aes(fill=rptec, xmin=x, filter=type=="gene")) +
overlay_raw_map("hsa03410", transparent_colors = c("#cccccc","#FFFFFF","#BFBFFF","#BFFFBF"))+
scale_fill_manual(values=c("steelblue","tomato"),
name="urothelial|rptec")+
theme_void()
gg2 <- gg + right + plot_layout(design="
AAAA###
AAAABBB
AAAABBB
AAAA###
"
)
gg2
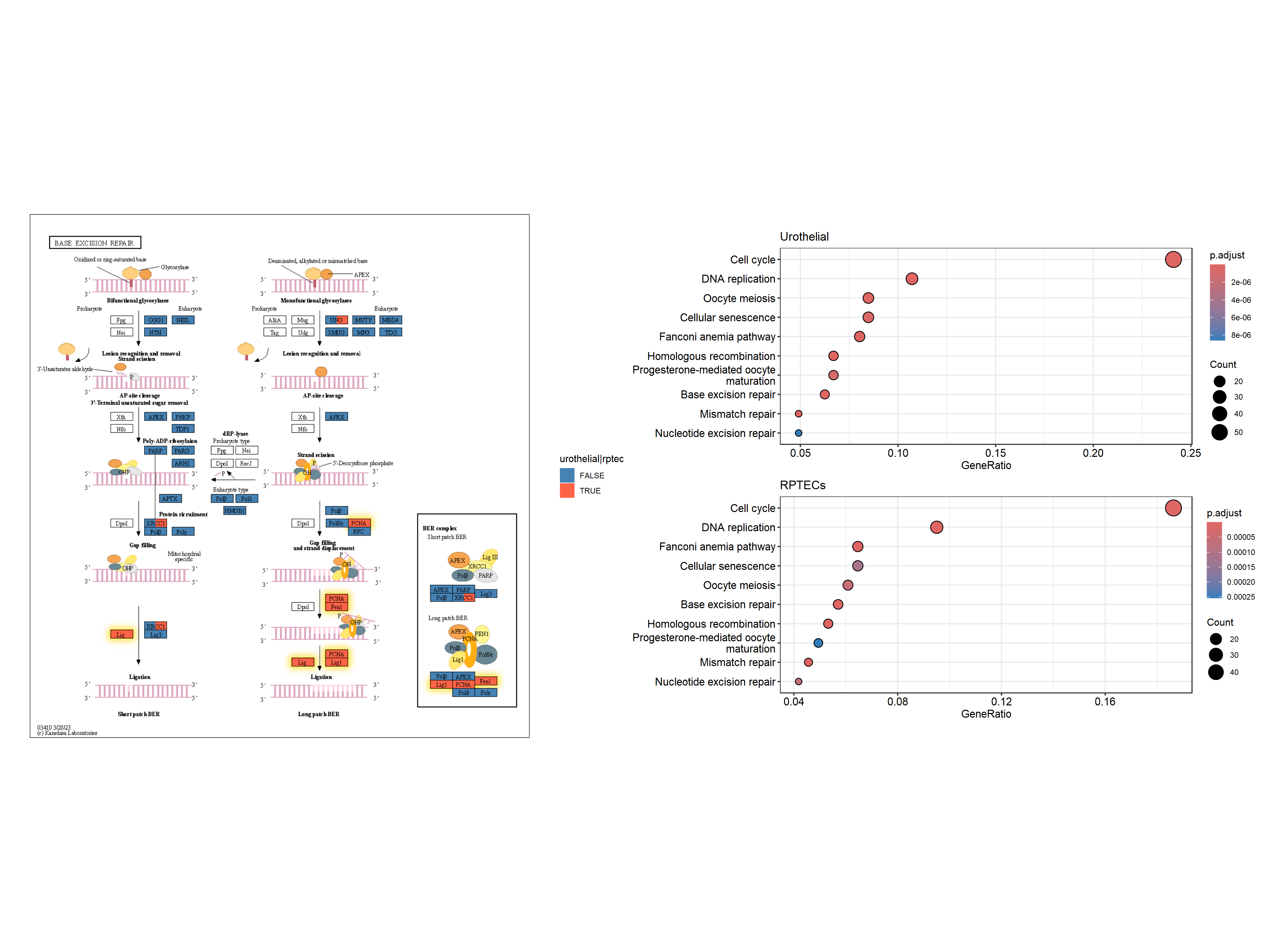
跨多个通路的多重富集分析结果
除了天然布局外,有时还可以在多个通路中显示有趣的基因,例如DEGs。在这里,我们使用散点图库来可视化跨多个途径的多个富集分析结果。
library(scatterpie)
## Obtain enrichment analysis results
entrezid <- uroUp |>
clusterProfiler::bitr("SYMBOL","ENTREZID",org.Hs.eg.db)
cp <- clusterProfiler::enrichKEGG(entrezid$ENTREZID)
entrezid2 <- gls$day3_up_rptec |>
clusterProfiler::bitr("SYMBOL","ENTREZID",org.Hs.eg.db)
cp2 <- clusterProfiler::enrichKEGG(entrezid2$ENTREZID)
## Filter to interesting pathways
include <- (data.frame(cp) |> row.names())[c(1,3,4)]
pathways <- data.frame(cp)[include,"ID"]
pathways
#> [1] "hsa04110" "hsa03460" "hsa03440"
我们获得多个通路数据(该函数返回原生坐标,但我们忽略它们)。
g1 <- multi_pathway_native(pathways, row_num=1)
g2 <- g1 |> mutate(new_name=
ifelse(name=="undefined",
paste0(name,"_",pathway_id,"_",orig.id),
name)) |>
convert(to_contracted, new_name, simplify=FALSE) |>
activate(nodes) |>
mutate(purrr::map_vec(.orig_data,function (x) x[1,] )) |>
mutate(pid1 = purrr::map(.orig_data,function (x) unique(x["pathway_id"]) )) |>
mutate(hsa03440 = purrr:::map_lgl(pid1, function(x) "hsa03440" %in% x$pathway_id) ,
hsa04110 = purrr:::map_lgl(pid1, function(x) "hsa04110" %in% x$pathway_id),
hsa03460 = purrr:::map_lgl(pid1, function(x) "hsa03460" %in% x$pathway_id))
nds <- g2 |> activate(nodes) |> data.frame()
eds <- g2 |> activate(edges) |> data.frame()
rmdup_eds <- eds[!duplicated(eds[,c("from","to","subtype_name")]),]
g2_2 <- tbl_graph(nodes=nds, edges=rmdup_eds)
g2_2 <- g2_2 |> activate(nodes) |>
mutate(
in_pathway_uro=append_cp(cp, pid=include,name="new_name"),
x=NULL, y=NULL,
in_pathway_rptec=append_cp(cp2, pid=include,name = "new_name"),
id=convert_id("hsa",name = "new_name")) |>
morph(to_subgraph, type!="group") |>
mutate(deg=centrality_degree(mode="all")) |>
unmorph() |>
filter(deg>0)
在这里,我们还将基于图的聚类结果分配给图,并缩放节点的大小,以便节点可以通过散点图可视化。
V(g2_2)$walktrap <- igraph::walktrap.community(g2_2)$membership
## Scale the node size
sizeMin <- 0.1
sizeMax <- 0.3
rawMin <- min(V(g2_2)$deg)
rawMax <- max(V(g2_2)$deg)
scf <- (sizeMax-sizeMin)/(rawMax-rawMin)
V(g2_2)$size <- scf * V(g2_2)$deg + sizeMin - scf * rawMin
## Make base graph
g3 <- ggraph(g2_2, layout="nicely")+
geom_edge_parallel(alpha=0.9,
arrow=arrow(length=unit(1,"mm")),
aes(color=subtype_name),
start_cap=circle(3,"mm"),
end_cap=circle(8,"mm"))+
scale_edge_color_discrete(name="Edge type")
graphdata <- g3$data
最后,我们用于可视化。背景散点表示基因是否在通路中,前景表示基因是否在多个数据集中差异表达。我们突出显示了在两个数据集中通过金色差异表达的基因。geom_scatterpie
g4 <- g3+
ggforce::geom_mark_rect(aes(x=x, y=y, group=walktrap),color="grey")+
geom_scatterpie(aes(x=x, y=y, r=size+0.1),
color="transparent",
legend_name="Pathway",
data=graphdata,
cols=c("hsa04110", "hsa03440","hsa03460")) +
geom_scatterpie(aes(x=x, y=y, r=size),
color="transparent",
data=graphdata, legend_name="enrich",
cols=c("in_pathway_rptec","in_pathway_uro"))+
ggfx::with_outer_glow(geom_scatterpie(aes(x=x, y=y, r=size),
color="transparent",
data=graphdata[graphdata$in_pathway_rptec & graphdata$in_pathway_uro,],
cols=c("in_pathway_rptec","in_pathway_uro")), colour="gold", expand=3)+
geom_node_point(shape=19, size=3, aes(filter=!in_pathway_uro & !in_pathway_rptec & type!="map"))+
geom_node_shadowtext(aes(label=id, y=y-0.5), size=3, family="sans", bg.colour="white", colour="black")+
theme_void()+coord_fixed()
g4
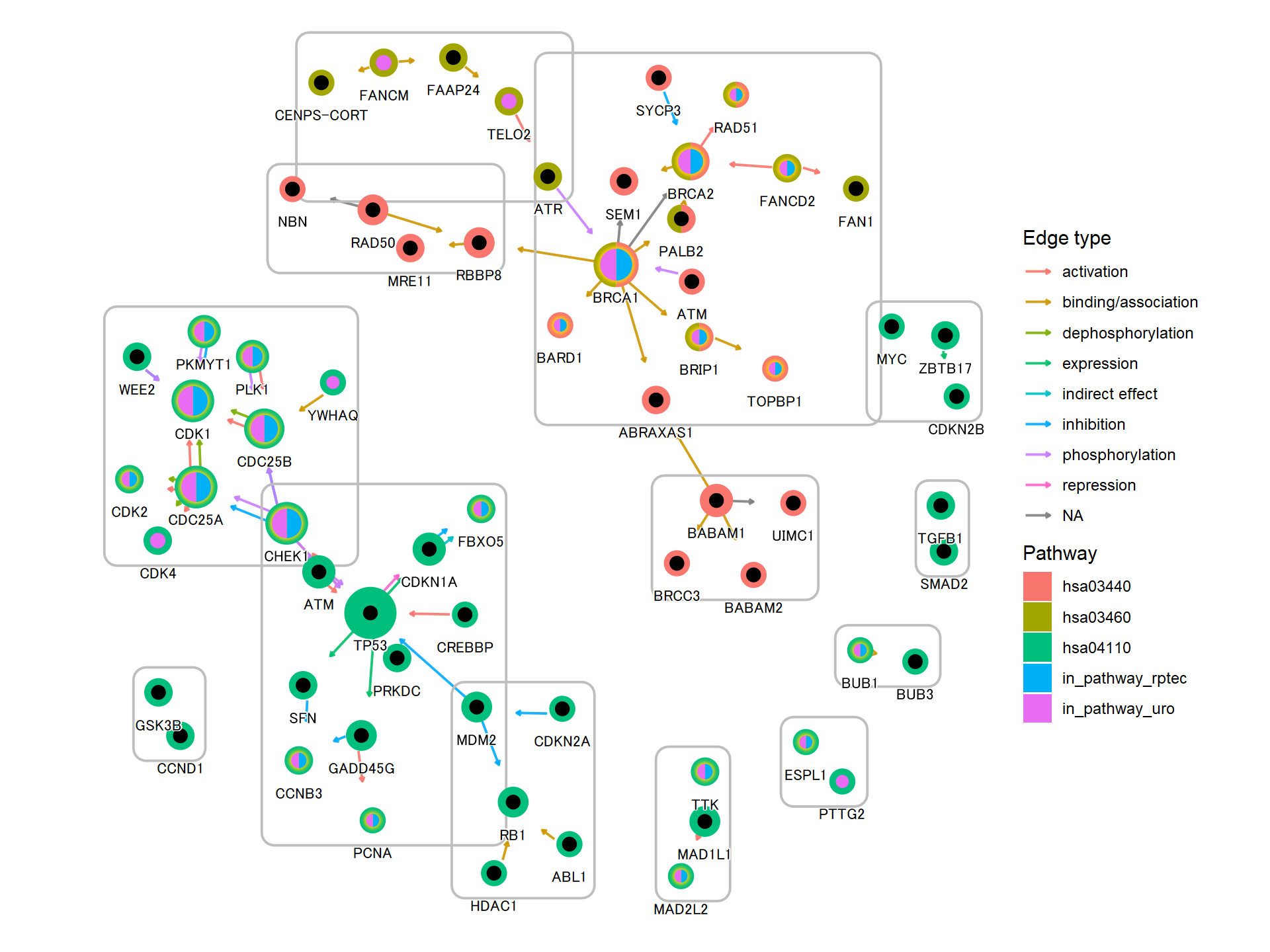
5.4 在KEGG图谱上投影基因调控网络
使用此软件包,可以将推断的网络(例如基因调控网络或由其他软件推断的 KO 网络)投射到 KEGG 图谱上。以下是使用 将 CBNplot 推断的通路内的 KO 网络子集投影到相应通路的参考图上的示例。当然,也可以投影使用其他方法创建的网络。MicrobiomeProfiler
library(dplyr)
library(igraph)
library(tidygraph)
library(CBNplot)
library(ggkegg)
library(MicrobiomeProfiler)
data(Rat_data)
ko.res <- enrichKO(Rat_data)
exp.dat <- matrix(abs(rnorm(910)), 91, 10) %>% magrittr::set_rownames(value=Rat_data) %>% magrittr::set_colnames(value=paste0('S', seq_len(ncol(.))))
returnnet <- bngeneplot(ko.res, exp=exp.dat, pathNum=1, orgDb=NULL,returnNet = TRUE)
pg <- pathway("ko00650")
joined <- combine_with_bnlearn(pg, returnnet$str, returnnet$av)
绘制生成的地图。在此示例中,估计的强度首先用彩色边缘显示,然后参考图的边缘在其顶部以黑色绘制。此外,两个图形中包含的边缘都以黄色突出显示。CBNplot
## Summarize duplicate edges including `strength` attribute
number <- joined |> activate(edges) |> data.frame() |> group_by(from,to) |>
summarise(n=n(), incstr=sum(!is.na(strength)))
## Annotate them
joined <- joined |> activate(edges) |> full_join(number) |> mutate(both=n>1&incstr>0)
joined |>
activate(nodes) |>
filter(!is.na(type)) |>
mutate(convertKO=convert_id("ko")) |>
activate(edges) |>
ggraph(x=x, y=y) +
geom_edge_link0(width=0.5,aes(filter=!is.na(strength),
color=strength), linetype=1)+
ggfx::with_outer_glow(
geom_edge_link0(width=0.5,aes(filter=!is.na(strength) & both,
color=strength), linetype=1),
colour="yellow", sigma=1, expand=1)+
geom_edge_link0(width=0.1, aes(filter=is.na(strength)))+
scale_edge_color_gradient(low="blue",high="red")+
geom_node_rect(color="black", aes(fill=type))+
geom_node_text(aes(label=convertKO), size=2)+
geom_node_text(aes(label=ifelse(grepl(":", graphics_name), strsplit(graphics_name, ":") |>
sapply("[",2) |> stringr::str_wrap(22), stringr::str_wrap(graphics_name, 22)),
filter=!is.na(type) & type=="map"), family="serif",
size=2, na.rm=TRUE)+
theme_void()
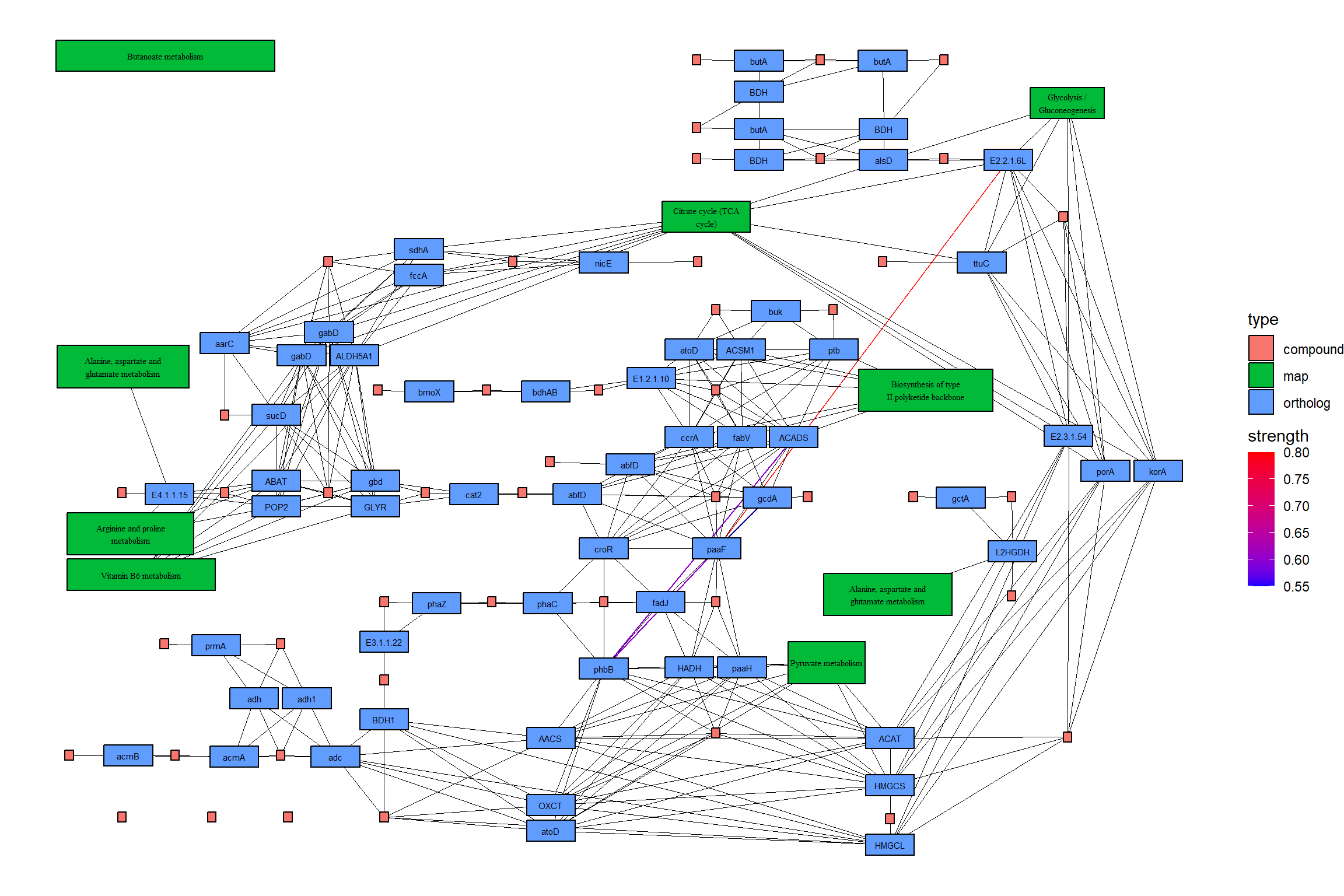
5.4.1 投影到原始 KEGG 地图上
您可以直接将推断网络投影到原始 PATHWAY 地图上,这样可以直接比较您自己的数据集中精选数据库和推断网络的知识。
raws <- joined |>
ggraph(x=x, y=y) +
geom_edge_link(width=0.5,aes(filter=!is.na(strength),
color=strength),
linetype=1,
arrow=arrow(length=unit(1,"mm"),type="closed"),
end_cap=circle(5,"mm"))+
scale_edge_color_gradient2()+
overlay_raw_map(transparent_colors = c("#ffffff"))+
theme_void()
raws
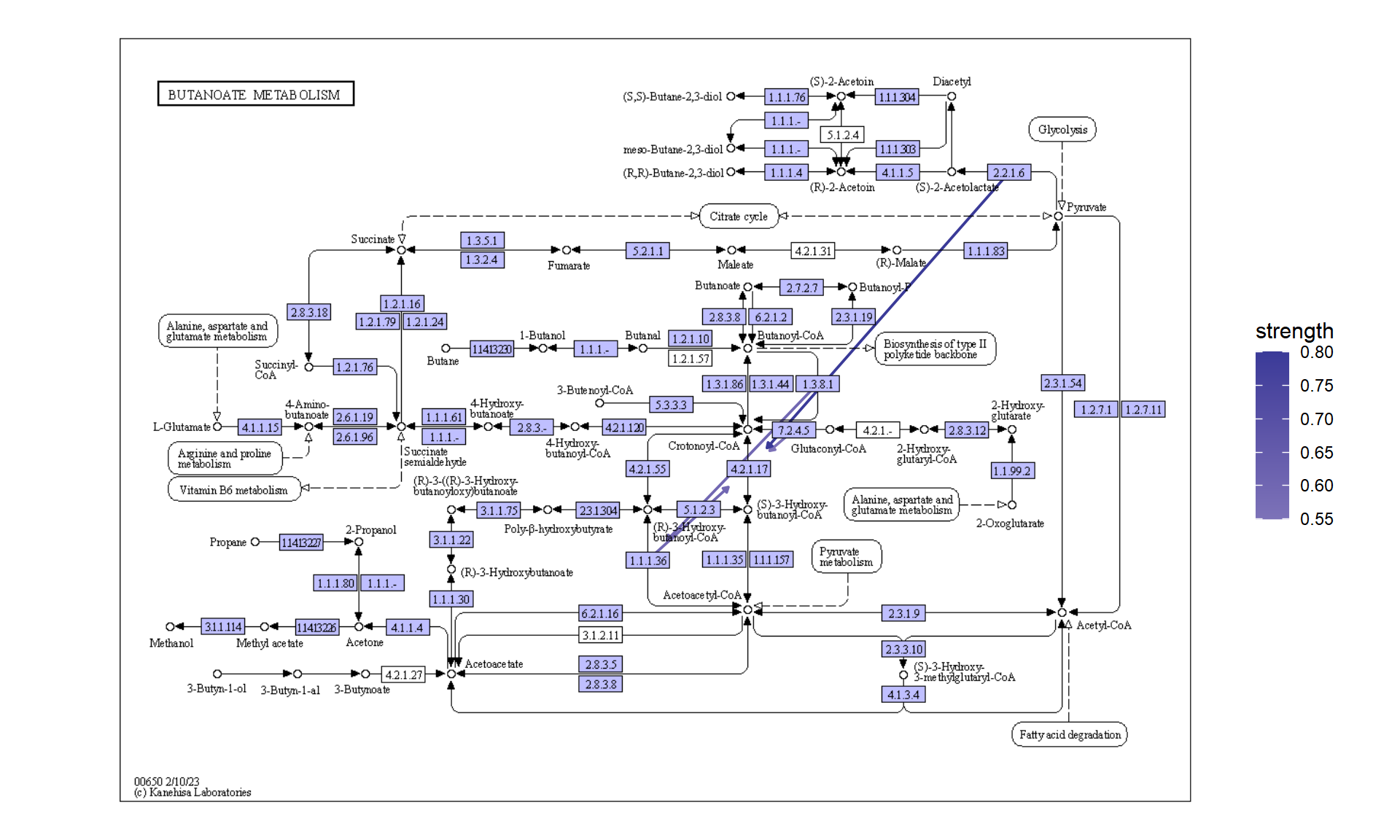
5.5 分析单细胞转录组学中的簇标记基因
该软件包也可应用于单细胞分析。例如,考虑将簇之间的标记基因映射到 KEGG 通路上,并将它们与降维图一起绘制。在这里,我们使用包。我们进行基本面分析。Seurat
library(Seurat)
library(dplyr)
# dir = "../filtered_gene_bc_matrices/hg19"
# pbmc.data <- Read10X(data.dir = dir)
# pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k",
# min.cells=3, min.features=200)
# pbmc <- NormalizeData(pbmc)
# pbmc <- FindVariableFeatures(pbmc, selection.method = "vst")
# pbmc <- ScaleData(pbmc, features = row.names(pbmc))
# pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
# pbmc <- FindNeighbors(pbmc, dims = 1:10, verbose = FALSE)
# pbmc <- FindClusters(pbmc, resolution = 0.5, verbose = FALSE)
# markers <- FindAllMarkers(pbmc)
# save(pbmc, markers, file="../sc_data.rda")
## To reduce file size, pre-calculated RDA will be loaded
load("../sc_data.rda")
随后,我们绘制了PCA降维的结果。
其中,在本研究中,我们对簇 1 和 5 的标记基因进行了富集分析。
library(clusterProfiler)
## Directly access slots in Seurat
pcas <- data.frame(
pbmc@reductions$pca@cell.embeddings[,1],
pbmc@reductions$pca@cell.embeddings[,2],
pbmc@active.ident,
pbmc@meta.data$seurat_clusters) |>
`colnames<-`(c("PC_1","PC_2","Cell","group"))
aa <- (pcas %>% group_by(Cell) %>%
mutate(meanX=mean(PC_1), meanY=mean(PC_2))) |>
select(Cell, meanX, meanY)
label <- aa[!duplicated(aa),]
dd <- ggplot(pcas)+
geom_point(aes(x=PC_1, y=PC_2, color=Cell))+
shadowtext::geom_shadowtext(x=label$meanX,y=label$meanY,label=label$Cell, data=label,
bg.colour="white", colour="black")+
theme_minimal()+
theme(legend.position="none")
marker_1 <- clusterProfiler::bitr((markers |> filter(cluster=="1" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
marker_5 <- clusterProfiler::bitr((markers |> filter(cluster=="5" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
mk1_enrich <- enrichKEGG(marker_1)
mk5_enrich <- enrichKEGG(marker_5)
从中获取颜色信息,并使用 获取通路。在这里,我们选择了 ,节点根据降维图中的颜色着色,两个聚类中的标记都按指定的颜色 () 着色。这促进了通路信息(如KEGG)与单细胞分析数据之间的联系,从而能够创建直观且易于理解的视觉表示。ggplot2``ggkegg``Osteoclast differentiation (hsa04380)``ggfx``tomato
## Make color map
built <- ggplot_build(dd)$data[[1]]
cols <- built$colour
names(cols) <- as.character(as.numeric(built$group)-1)
gr_cols <- cols[!duplicated(cols)]
g <- pathway("hsa04380") |> mutate(marker_1=append_cp(mk1_enrich),
marker_5=append_cp(mk5_enrich))
gg <- ggraph(g, layout="manual", x=x, y=y)+
geom_node_rect(aes(filter=marker_1&marker_5), fill="tomato")+ ## Marker 1 & 5
geom_node_rect(aes(filter=marker_1&!marker_5), fill=gr_cols["1"])+ ## Marker 1
geom_node_rect(aes(filter=marker_5&!marker_1), fill=gr_cols["5"])+ ## Marker 5
overlay_raw_map("hsa04380", transparent_colors = c("#cccccc","#FFFFFF","#BFBFFF","#BFFFBF"))+
theme_void()
gg+dd+plot_layout(widths=c(0.6,0.4))
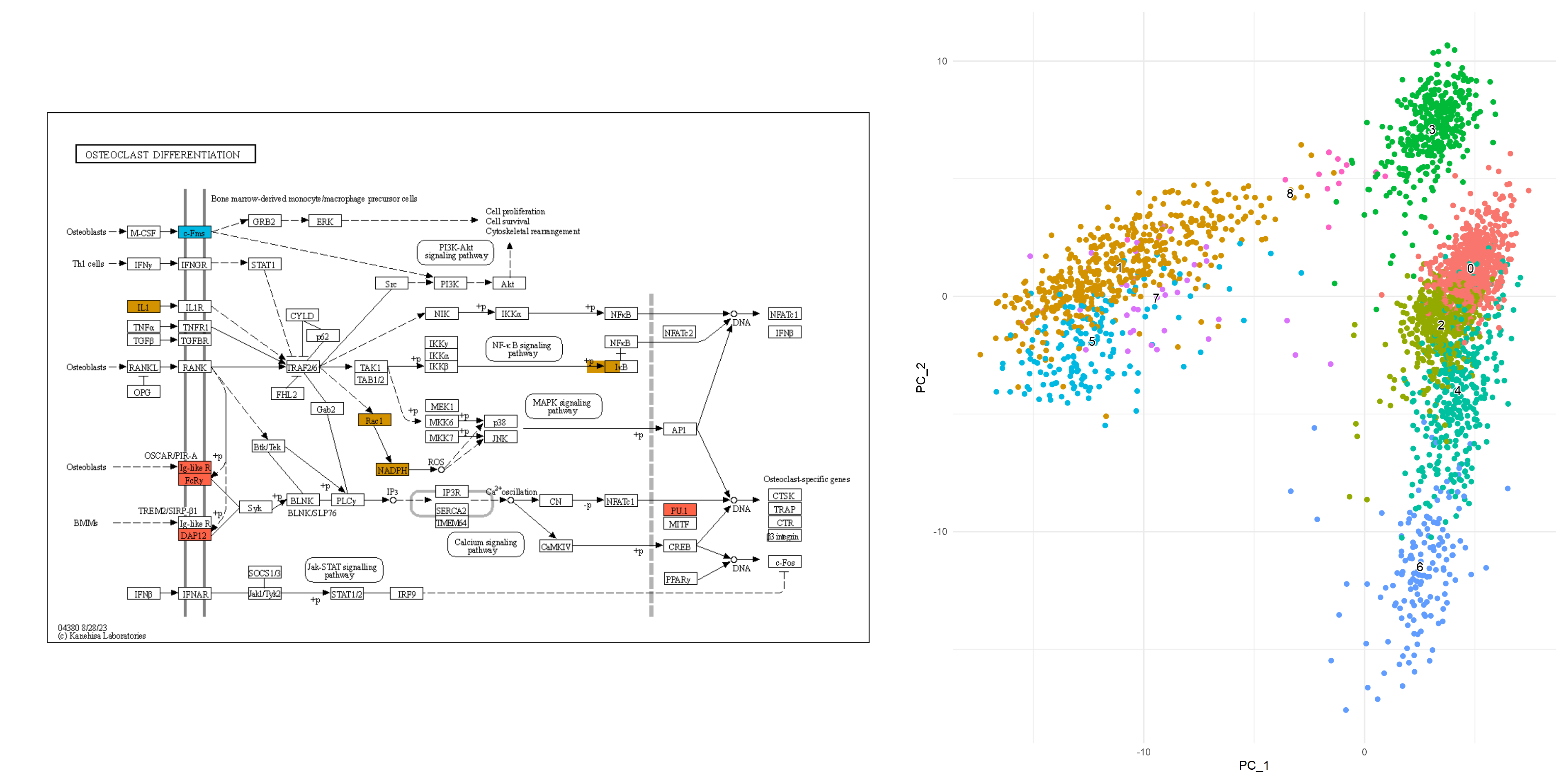
5.5.1 组成多个通路的示例
我们可以在多种途径中检查标记基因,以更好地了解标记基因的作用。
library(clusterProfiler)
library(org.Hs.eg.db)
subset_lab <- label[label$Cell %in% c("1","4","5","6"),]
dd <- ggplot(pcas) +
ggfx::with_outer_glow(geom_node_point(size=1,
aes(x=PC_1, y=PC_2, filter=group=="1", color=group)),
colour="tomato", expand=3)+
ggfx::with_outer_glow(geom_node_point(size=1,
aes(x=PC_1, y=PC_2, filter=group=="5", color=group)),
colour="tomato", expand=3)+
ggfx::with_outer_glow(geom_node_point(size=1,
aes(x=PC_1, y=PC_2, filter=group=="4", color=group)),
colour="gold", expand=3)+
ggfx::with_outer_glow(geom_node_point(size=1,
aes(x=PC_1, y=PC_2, filter=group=="6", color=group)),
colour="gold", expand=3)+
shadowtext::geom_shadowtext(x=subset_lab$meanX,
y=subset_lab$meanY, label=subset_lab$Cell,
data=subset_lab,
bg.colour="white", colour="black")+
theme_minimal()
marker_1 <- clusterProfiler::bitr((markers |> filter(cluster=="1" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
marker_5 <- clusterProfiler::bitr((markers |> filter(cluster=="5" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
marker_6 <- clusterProfiler::bitr((markers |> filter(cluster=="6" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
marker_4 <- clusterProfiler::bitr((markers |> filter(cluster=="4" & p_val_adj < 1e-50) |>
dplyr::select(gene))$gene,fromType="SYMBOL",toType="ENTREZID",OrgDb = org.Hs.eg.db)$ENTREZID
mk1_enrich <- enrichKEGG(marker_1)
mk5_enrich <- enrichKEGG(marker_5)
mk6_enrich <- enrichKEGG(marker_6)
mk4_enrich <- enrichKEGG(marker_4)
g1 <- pathway("hsa04612") |> mutate(marker_4=append_cp(mk4_enrich),
marker_6=append_cp(mk6_enrich),
gene_name=convert_id("hsa"))
gg1 <- ggraph(g1, layout="manual", x=x, y=y)+
overlay_raw_map("hsa04612", transparent_colors = c("#FFFFFF", "#BFBFFF", "#BFFFBF"))+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_4&marker_6), fill="white"),
colour="gold")+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_4&!marker_6), fill="white"),
colour=gr_cols["4"])+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_6&!marker_4), fill="white"),
colour=gr_cols["6"], expand=3)+
overlay_raw_map("hsa04612", transparent_colors = c("#B3B3B3", "#FFFFFF", "#BFBFFF", "#BFFFBF"))+
theme_void()
g2 <- pathway("hsa04380") |> mutate(marker_1=append_cp(mk1_enrich),
marker_5=append_cp(mk5_enrich))
gg2 <- ggraph(g2, layout="manual", x=x, y=y)+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_1&marker_5),
fill="white"), ## Marker 1 & 5
colour="tomato")+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_1&!marker_5),
fill="white"), ## Marker 1
colour=gr_cols["1"])+
ggfx::with_outer_glow(
geom_node_rect(aes(filter=marker_5&!marker_1),
fill="white"), ## Marker 5
colour=gr_cols["5"])+
overlay_raw_map("hsa04380",
transparent_colors = c("#cccccc","#FFFFFF","#BFBFFF","#BFFFBF"))+
theme_void()
left <- (gg2 + ggtitle("Marker 1 and 5")) /
(gg1 + ggtitle("Marker 4 and 6"))
final <- left + dd + plot_layout(design="
AAAAA###
AAAAACCC
BBBBBCCC
BBBBB###
")
final
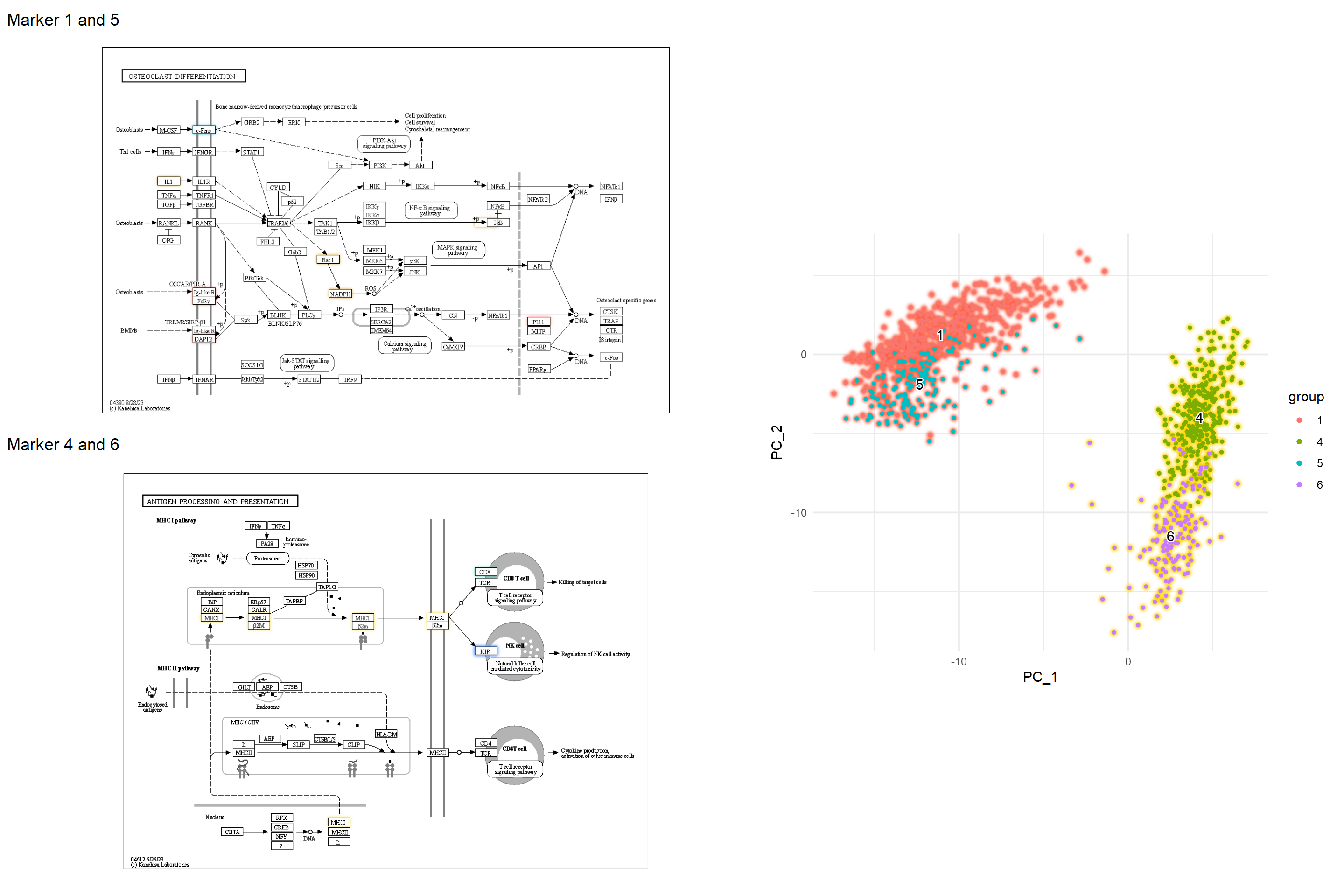
5.5.2 原始地图上数值的条形图
对于它们在多个聚类中丰富的节点,我们可以绘制数值的条形图。引用的代码由 inscaven 提供。
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
“hsa04380”,
transparent_colors = c(“#cccccc”,“#FFFFFF”,“#BFBFFF”,“#BFFFBF”))+
theme_void()
left <- (gg2 + ggtitle(“Marker 1 and 5”)) /
(gg1 + ggtitle(“Marker 4 and 6”))
final <- left + dd + plot_layout(design="
AAAAA###
AAAAACCC
BBBBBCCC
BBBBB###
")
final

#### 5.5.2 原始地图上数值的条形图
对于它们在多个聚类中丰富的节点,我们可以绘制数值的条形图。引用的代码由 inscaven [提供]( )。
**网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。**
**[需要这份系统化的资料的朋友,可以点击这里获取!](https://bbs.csdn.net/forums/4f45ff00ff254613a03fab5e56a57acb)**
**一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!**


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









