







抽象
背景
生物学大数据的出现为获得生物学见解带来了机遇,但也带来了挑战。对于玉米,已经生成了大量公开可用的转录组数据集,但缺乏全面的分析。
结果
我们使用大量的 RNA-seq 数据,基于图形高斯模型构建了一个玉米基因共表达网络。该网络包含 20,269 个基因,组装成 964 个基因模块,这些模块在各种植物过程中发挥作用,例如细胞组织、花序、叶子和籽粒的发育、营养物质(例如氮和磷酸盐)的吸收和利用、苯并氧杂氮苷、氧磷脂、类黄酮和蜡的代谢,以及对压力的反应。其中,花序发育模块富含控制植物结构和内核结构的驯化基因 (如 ra1、ba1、gt1、tb1、tga1),而其他多个模块与不同的农艺性状相关。这些模块中包含的转录因子充当相同模块中基因的已知或潜在表达调节因子,表明它们是相关生物过程的候选调节因子。与已建立的拟南芥网络进行比较,揭示了参与细胞组织、营养吸收和利用以及代谢的特定模块的保守基因关联模式。该分析还确定了两个物种之间在协调发育途径的模块方面的显着差异。
结论
该网络阐明了在进化分化的背景下不同物种之间的基因模块是如何组织的,并突出了其结构和基因内容可以为具有应用潜力的玉米基因功能研究提供重要资源的模块。
背景
过去二十年的进步产生了大量的转录组数据集。越来越完整的转录组数据可以与基因和基因组结构合并和整合。它们提供了各种条件下生物体内基因表达动力学的无偏快照。虽然细胞、器官或条件特异性表达谱比比皆是,但推断控制和产生观察到的基因表达动力学的潜在基因调控回路仍然是一个关键挑战。为了解决这个问题,基因网络分析已成为一种可以过滤和优化大型基因表达数据集分析的工具。这种基因网络由基因(节点)和基因之间的连接(边缘)组成,这些基因代表表达数据背后的共表达动力学或关联模式。根据“关联罪恶”范式,连接的基因可能具有相似的功能,是同一复合物或通路的一部分,或参与相同的信号回路 [1]。基因网络可以根据其关联基因揭示的功能为未知基因分配推定的功能,或者为现有途径识别新基因 [2, 3]。
与随机网络不同,基因网络通常识别基因模块,对高度互连的基因组进行分类和追踪,这些基因具有相似的表达模式,并且通常可以通过它们与特定生物过程中的功能的关系来识别。每个模块都可以看作是控制一个或多个生物功能的单元。因此,模块识别并汇集了生物系统的片段。基因网络分析已被用于检测与人类疾病或植物种子萌发等相关的基因模块,并研究酵母、植物和动物的转录组景观和基因模块组织[4,5,6,7,8,9,10]。
来自微阵列数据的基因网络已被描述为拟南芥、玉米和水稻等植物物种[6,8,11,12,13]。 这些网络是通过多种方法构建的,这些方法采用不同的方法来测量基因之间的相互作用。最常见的是利用 Pearson 相关系数 (PCC) 来测量基因之间表达相似性的共表达网络,其中 PPC 大于所选阈值的基因对被认为相互相互作用。例子包括两个拟南芥网络:一个确定了参与光合作用、维生素代谢或细胞周期调节等过程的基因簇,而第二个网络揭示了与细胞器和组织特异性功能的分组[8,11]。水稻和玉米网络也通过加权基因共表达网络分析(WGCNA)方法组装,该方法利用PCC的幂函数来评估表达相似性[6,12,13,14]。具体来说,Downs 等人使用 WGCNA 构建了一个玉米发育基因共表达网络,该网络捕获了具有组织和发育阶段特异性的模块,而 Ficklin 和 Feltus 比较了玉米和水稻的 WGCNA 网络,以识别保守的模块[6,13]。
另一种进行共表达网络分析的方法使用图形高斯模型 (GGM),该模型利用部分相关 (Pcor) 来识别基因之间的关联关系 [15,16,17]。值得注意的是,Pcor 确定了在去除所有其他基因的影响后两个基因之间仍然存在的相关性。Pcor 测量基因之间的直接关联,而 PCC 通常无法区分直接和间接关联 [15, 16]。因此,Pcor 被认为是比 PCC 更好的基因网络分析指标 [16, 18]。然而,GGM 的利用受到样本数量必须远大于基因数量的要求所阻碍。最初的设计只能计算几千个基因中的 Pcor 数据 [16]。以前,我们开发了一种基于随机采样的方法来克服这一障碍,并构建了第一个用于拟南芥的全基因组GGM网络,随后又构建了一个名为AtGGM2014的更新网络模型[7,17]。与其他网络相比,AtGGM2014 包含更多的基因,并确定了参与多种植物过程的额外模块,如发育、代谢、对压力的反应和对激素的反应 [7]。例如,在许多人信息丰富的基因模块中,它包括生长素、脱落酸、茉莉酸、赤霉素、细胞分裂素、乙烯和水杨酸等植物激素的激素信号模块,证明了该网络促进拟南芥基因功能系统生物学研究的潜力。
最近,不同小组生成了大量基于 RNA-Seq 的玉米基因表达数据集,并存放在公共领域。这些数据集包括79个玉米组织的基因表达图谱[19,20],以及特定器官的表达数据集,如花序[21]、叶子[22]、小叶[23]、胚胎和胚乳[24],以及胚乳中不同区室的转录组数据集[25,26]。 其他研究是通过监测玉米对非生物胁迫的反应[27,28]、真菌感染[29]和不同的营养制度[30]得出的。这些数据集为玉米功能基因组学研究提供了极其有价值的信息。根据 23 个不同组织的表达数据构建了一个专注于发育的基于玉米 RNA-Seq 的基因网络,通过 WGCNA 方法鉴定了 19 个基因模块 [31]。然而,据我们所知,到目前为止还缺少一个全面的基因网络分析,它结合了许多不同玉米数据集的财富,并将它们合并成一个包容性网络,以概述未来的研究机会。
在这里,我们使用来自 NCBI SRA 数据库中保存的 787 个 RNA-Seq 运行的表达数据构建了一个玉米 GGM 基因共表达网络。从该网络中鉴定出 964 个基因模块,突出了各种细胞组织、发育、营养、代谢和应激反应途径中的功能。作为示例,我们详细描述了涉及花序、叶茎和籽粒发育、氮和磷酸盐的吸收和利用、苯并恶氮菌、氧磷脂、类黄酮和蜡的代谢,以及对热应激、内质网 (ER) 应激和真菌感染的反应的模块。这些模块为相关的生物过程提供了一般情况,并为未来的功能研究确定了已知基因以及潜在的、迄今为止尚未连接的候选基因。重要的是,其中许多模块包含转录因子基因,这些基因充当相同模块内基因的潜在基因表达调节因子。此外,该玉米网络与以前发表的拟南芥网络进行了比较[7]。这种并置揭示了两个物种的保守模块和发散模块。鉴定出的基因模块进一步用于分析玉米叶片发育系列的数据集[22],证明了该网络在系统生物学分析中的有用性。
结果
玉米 GGM 基因网络概述
使用存储在 NCBI 序列读取档案 (SRA) 数据库中的玉米 RNA-Seq 转录组数据集构建基于 GGM 的玉米基因共表达网络。下载公开可用的原始数据文件 (.sra),与玉米参考基因组 (AGP_v3.22) 进行映射,并处理成基因表达值。在去除标测率为 <70%) 的文件后,保留了 36 项不同研究(附加文件 1:表 S1)的 787 个 RNA-Seq 运行,并将其每百万张映射读数中转录本的每千碱基片段数 (FPKM) 标准化基因表达值合并到一个大型基因表达矩阵中。这些数据集监测来自各种组织和发育阶段、各种生物和非生物胁迫处理后的玉米转录组,或来自特征明确的突变体。过滤掉具有最大 FPKM 值 <20 的基因,得到一个基因表达矩阵,其中包括 29,316 个玉米基因(行)和 787 个 RNA-Seq 运行(列)。与其他RNA-Seq数据类似,这些玉米RNA-Seq数据显示出均值-方差依赖性[32],即平均表达值较高的基因更有可能具有较大的方差。通过对数变换转换基因表达矩阵以减少这种均值-方差依赖性(附加文件 2:图 S1),使数据集更适合相关性分析 [32]。以前曾使用对数转化进行基因共表达和基因聚类分析,使用RNA-Seq数据[33,34,35,36,37,38,39,40]。然后,按照前面描述的程序,使用对数转换的基因表达矩阵计算偏相关系数 (Pcor) [17]。图 1a 显示了 Pcors 在所有基因对之间的分布,其中 98.4% 的基因对在 -0.01 和 0.01 之间。该基因与 |Pcor|保留 > = 0.035 (pValue = 2.22E-16) 用于网络构建。此外,还计算了所有基因对的 Pearson 相关系数 (PCC) 以及 |PCC|< 0.35 被去除,理由是低 PCC 很可能表明基因之间的独立性。结果,选择了 123,093 个基因对,Pcor > = 0.035 和 PCC > = 0.35,573 个基因对,Pcor <= -0.035 和 PCC < = -0.35 的基因对被选择用于基因网络构建(附加文件 3:表 S2)。
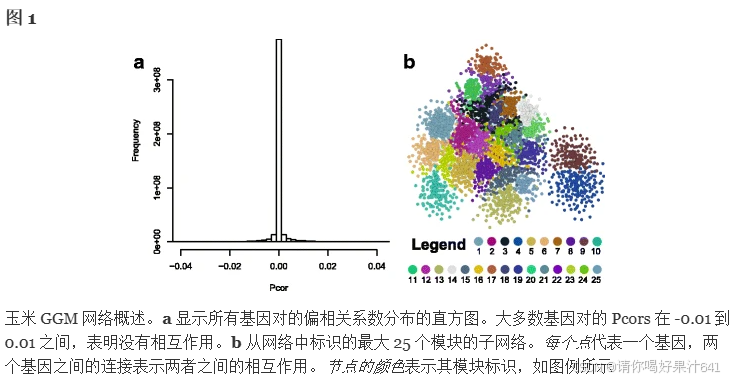
这一推理导致了玉米 GGM 网络,包括 20,269 个基因和 123,666 个共表达基因对 (占所有可能的基因对的 0.06%)。该网络的聚类系数 (C) 为 0.209,而相同大小的随机网络的预期 C 仅为 0.0006 [41]。由于 C 衡量网络的潜在模块化 [42],玉米 GGM 网络的大 C 表示模块化的高度特性。事实上,使用 MCL 程序 [43] 的网络聚类程序确定了 964 个基因模块,每个模块包含 5 个或更多基因,这 964 个模块总共包含 16,668 个基因。这些模块可以被视为共表达模块,其基因具有相似的共表达模式,并且根据“关联罪恶感”范式,可能具有相似的功能或参与相同的通路。其余 3601 个基因被分配到具有 4 个或更少基因的小模块,这些模块在进一步分析中没有考虑。在我们的网络中,最大的 222 个模块包含 15 到 306 个基因(图 D)。1b,附加文件 4:表 S3)。根据基因本体论 (GO) 富集分析,其中许多模块分为 5 个不同的类别,其功能包括:i. 细胞组织;ii. 发展;第三营养吸收和利用;iv. 新陈代谢,包括初级和次级新陈代谢;和v. 压力反应(表1和附加文件1:表S4)。细胞组织和初级代谢的模块包括用于细胞周期调节的模块(模块 #60)、DNA 复制 (#64)、细胞骨架组织 (#69)、核小体组装 (#49)、光合作用(#20 和 129)和线粒体相关功能(#14 和 86)(表 1)。与已发表的拟南芥网络 AtGGM2014 [7] 中报道的类似模块相比,其中一些模块是高度保守的。
方法
玉米 RNA-Seq 数据采集
分析中使用了存放在 NCBI SRA 数据库中的公开可用的玉米 RNA-Seq 转录组数据集。这些数据集按研究组织。对这些研究进行人工检查,以过滤掉那些专注于非编码 RNA 的研究或那些测量大量玉米品种中相同组织转录组的研究。还删除了 RNA-Seq 运行少于 5 次或未发表文章的研究。结果,保留了 36 项研究并下载了其原始数据文件 (sra)。对于 RNA-Seq 数据处理,使用 FASTX-toolkit 流程版本 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/) 从原始序列读数中删除接头序列(如果存在)。使用 FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) 检查序列质量,并通过 FASTX-toolkit 过滤低质量读数。然后使用默认设置的 Tophat v2.0.10 [97] 将剩余的读数映射到玉米基因组 AGP v3.22 (Ensembl Plants, http://plants.ensembl.org)。在去除定位率小于 70% 的文件后,通过 Cufflinks v2.1.1 分析来自 787 次 RNA-Seq 运行的 bam 文件以获得基因表达值 (FPKM) [98]。
玉米 GGM 网络建设
然后将基因表达数据合并为具有 787 列的单个基因表达矩阵,并过滤掉低表达基因(所有样品中的最大 FPKM 值小于 20),得到具有 29,316 个基因和 787 列的矩阵。通过对数对矩阵进行对数转换 [32,33,34,35,36,37,38,39,40]2(FPKM + 1) 函数,该过程显著降低了数据集的均值-方差依赖性(附加文件 2:图 S1)。然后,按照前面描述的方法,使用对数转换的基因表达矩阵进行部分相关计算 [17]。简而言之,该计算涉及一个具有 25,000 次迭代的过程。在每次迭代中,随机选择 2000 个基因,并通过 R 中 GeneNet v1.2.13 包中的“ggm.estimate.pcor”函数估计基因对之间的偏相关系数 [16]。Pcors 在每次迭代中都被记录下来。经过 25,000 次迭代后,对于每个基因对,选择绝对值最低的 Pcor 作为其最终 Pcor。还计算了所有基因对之间的 PCC。选择 Pcor > = 0.035 且 PCC > = 0.35 的基因对以及 Pcor <= -0.035 且 PCC < = -0.35 的基因对用于基因网络构建(附加文件 2:表 S2),从而产生基于对数转换基因表达数据的玉米 GGM 基因网络。
为了评估对数转换对网络质量的影响,还直接从基因表达矩阵构建了另一个基因网络,其原始 FPKM 值没有数据转换,指定为未转换的 FPKM 网络,保持所有其他参数与上述基于对数转换的网络相同。然后通过 R 中的 EGAD 包评估和比较这两个网络连接具有共享 GO 术语的玉米基因的能力 [99]。结果表明,基于对数变换的网络优于未变换的 FPKM 网络(附加文件 8:图 S9)。此外,基于对数转化的网络识别出未被未转化的FPKM网络恢复的基因模块,例如与ER应激反应(#95)和硝酸盐反应与同化(#72)相关的基因模块(数据未显示)。这些模块仅由基于对数转化的网络识别,其中包含在不同分析中被鉴定为与所讨论模块相关的基因 [73, 74, 91, 92]。
因此,我们考虑了基于对数变换的网络的更高功效,并且仅进一步分析和讨论了使用基于对数变换的网络(称为玉米 GGM 网络)的结果。
基因网络特性和基因模块鉴定
RBGL v 1.44.0 (http://bioconductor.org/packages/RBGL/) 的 R 包用于计算玉米 GGM 网络的聚类系数。通过 MCL 聚类算法对网络进行聚类,使用这些参数 “-I 1.5 -Scheme 7” [43]。然后通过 GOStats [100] 分析每个模块内的基因本体富集,并从 Gramene 数据库 (ftp://ftp.gramene.org/) 下载GO注释文件。玉米基因及其拟南芥同源物进一步用来自玉米GDB、TAIR和PlnTFDB的注释文件进行注释[101,102,103]。还通过二项分布测试了选定的模块的启动子基序富集。开发了一个 R 脚本,包含在随附的程序 MaizeGGM2016 中,用于提取基因模块的子网络,并使用来自已发表数据集的表达数据为选定模块中的基因绘制发育热图 [19,20,21, 23,24,25,26]。 分别使用 BioLayout Express 3D 和 Cytoscape 3.3 对整个 GGM 网络和提取的子网络进行布局和可视化 [104, 105]。
玉米网络和拟南芥网络之间的基因网络比较
为了能够比较玉米GGM网络和拟南芥网络AtGGM2014,使用InParanoid程序(v 4.1)[106]来识别拟南芥中最相似的玉米基因(如果存在)。对于玉米网络中的任何基因,如果拟南芥 AtGGM2014 网络中存在同源基因,则玉米基因在玉米网络内的紧邻基因被提取为 A 组。作为 B 组,还提取了拟南芥网络内其同源基因的邻居。如果 A 组中的任何基因在 B 组中具有同源基因,则原始玉米基因被认为在拟南芥网络内具有保守的相互作用。对于任何给定的模块,具有保守相互作用的基因的百分比被计算为进化保守性或分歧的指标。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









