







部分1:ChIP-seq的概念与用途
- ChIP-seq的定义:染色质免疫共沉淀测序的原理和过程。
- ChIP-seq的应用:
- 揭示基因调控机制(如转录因子结合位点)。
- 研究疾病发生机制(如癌症相关基因的异常调控)。
- 药物研发靶点发现(如增强子活性和基因表达调控)。
- 常见的研究类型:
- 转录因子结合位点研究。
- 组蛋白修饰研究(如H3K27me3、H3K4me3)。
- 染色质状态研究(如多梳蛋白、染色质重塑因子)。
- 增强子活性与基因调控。
部分2:ChIP-seq实验流程
- 实验步骤:
- 细胞固定(使用甲醛等交联剂)。
- 染色质剪切(超声波或酶解)。
- 抗体免疫共沉淀(特异性识别目标蛋白)。
- DNA逆交联与纯化。
- 构建测序文库。
- 高通量测序(Illumina平台)。
- 数据特点:
- 峰(Peak)信号:结合位点区域的DNA片段堆叠。
- 正负链(Strand)分布:显示结合事件的位置特征。
部分3:数据分析流程
- 质量控制:
- FastQC 检查测序质量。
- 去接头和低质量数据(如用Trimmomatic)。
- 序列比对(Mapping):
- 使用Bowtie2或BWA将序列比对到参考基因组。
- 生成BAM文件。
- 峰值识别(Peak Calling):
- 工具:MACS2。
- 使用对照样本校正背景噪声。
- 峰值注释:
- 注释工具:Homer 或 ChIPseeker。
- 注释到基因启动子、增强子等功能区域。
部分4:高级分析
- 差异结合分析(DBA):
- 比较不同条件下结合峰值的差异。
- 工具:DiffBind 或 csaw。
- 基序分析(Motif Analysis):
- 识别结合位点的核心序列模式(如转录因子的特定结合位点)。
- 工具:Homer 或 MEME。
- 功能富集分析:
- 基因本体(GO)分析。
- 通路富集(KEGG)。
部分5:可视化与整合
- 可视化工具:
- IGV:基因组浏览器。
- deepTools:生成信号热图或Profile图。
- 结果解读:
- 峰值堆叠显示结合强度。
- 分析染色质结构与调控机制。
部分6:组蛋白修饰与染色质状态
- 组蛋白修饰:
- 修饰类型:乙酰化、甲基化、磷酸化等。
- 表观遗传调控机制(如H3K27me3沉默基因)。
- 染色质状态注释:
- 使用无监督学习分析染色质区域(如隐马尔科夫模型)。
- 工具:ChromHMM 或 EpiCompare。
部分7:ChIP-seq的挑战与改进
- 挑战:
- 数据噪声与批次效应。
- 峰值识别精度问题。
- 改进:
- 数据去噪(如ChromImpute)。
- 插补缺失数据(如PREDICTD)。
部分1:ChIP-seq的概念与用途。
1. ChIP-seq的定义
ChIP-seq,全称是染色质免疫共沉淀测序(Chromatin Immunoprecipitation followed by Sequencing),是一种结合生物化学与基因组学的技术,用于研究蛋白质与DNA的相互作用。
- 核心原理:
ChIP-seq通过利用特异性抗体识别并捕获DNA上结合的特定蛋白质(如转录因子或修饰后的组蛋白),然后提取这些蛋白结合的DNA片段并进行测序分析,最终定位这些结合事件在整个基因组中的分布。
2. ChIP-seq的用途
2.1 揭示基因调控机制
- 通过定位特定转录因子或调控蛋白在基因组中的结合位点,可以分析它们如何调控基因表达。
- 例如:
- 分析p53在癌症细胞中的结合位点,揭示其在肿瘤抑制中的作用。
2.2 研究表观遗传学调控
组蛋白知识看这个:https://blog.csdn.net/u011262253/article/details/109957163?ops_request_misc=%257B%2522request%255Fid%2522%253A%25220dab189fb37970a636ede979c1e39ca8%2522%252C%2522scm%2522%253A%252220140713.130102334…%2522%257D&request_id=0dab189fb37970a636ede979c1e39ca8&biz_id=0&utm_medium=distribute.pc_search_result.none-task-blog-2allsobaiduend~default-1-109957163-null-null.142v100pc_search_result_base1&utm_term=%E7%BB%84%E8%9B%8B%E7%99%BD%E4%BF%AE%E9%A5%B0&spm=1018.2226.3001.4187
- 分析组蛋白修饰(如H3K27me3、H3K4me3)在基因组中的分布,探讨这些修饰如何影响基因表达。
- 例如:
- H3K27me3的富集与基因沉默相关,帮助解释细胞命运决定的分子机制。
2.3 疾病研究
- 功能异常:通过ChIP-seq找到与疾病发生相关的基因调控异常。
- 癌症研究:研究异常转录因子的结合位点,发现潜在的致病基因或调控网络。
2.4 药物开发
- 通过识别关键的基因调控位点,为药物研发提供靶点。
- 例如:
- 研究活跃增强子如何调控重要基因,为开发靶向调控药物提供线索。
3. ChIP-seq研究的主要类型
3.1 转录因子结合位点研究
- 目的:分析转录因子在基因组中的结合位点,构建调控网络。
- 靶标:特定的转录因子(如p53、c-Myc)。
- 应用:
- 研究细胞特异性基因表达调控机制。
- 探索转录因子在癌症、代谢病中的作用。
- 示例:
使用ChIP-seq研究肿瘤抑制因子p53的基因组结合位点,揭示其调控肿瘤抑制基因的机制。
3.2 组蛋白修饰研究
- 目的:探索特定组蛋白修饰(如H3K27me3、H3K4me3)在基因组中的分布,研究表观遗传调控。
- 靶标:修饰后的组蛋白。
- 应用:
- 研究表观遗传调控与细胞分化、疾病的关系。
- 示例:
使用ChIP-seq研究H3K4me3在启动子区域的分布,分析其如何激活基因表达。
3.3 染色质状态研究
- 目的:通过染色质重塑因子或修饰蛋白研究染色质的开放或紧密状态。
- 靶标:染色质重塑因子(如BRG1)。
- 应用:
- 分析染色质开放状态对基因表达的影响。
- 探究染色质结构在发育和疾病中的功能。
3.4 RNA聚合酶II结合位点研究
- 目的:研究RNA聚合酶II的结合位点,探索基因转录起始和延伸机制。
- 靶标:RNA聚合酶II(Pol II)。
- 应用:
- 分析基因转录的活跃区域。
- 示例:
使用ChIP-seq研究特定基因在细胞激活过程中的转录活性。
3.5 增强子活性研究
- 目的:鉴定基因组中活跃的增强子,了解其如何通过与转录因子结合调控基因表达。
- 靶标:增强子相关的组蛋白修饰(如H3K27ac)。
- 应用:
- 识别活跃增强子,研究基因表达的远程调控机制。
- 示例:
使用ChIP-seq检测H3K27ac标记,寻找发育基因的活跃增强子。
3.6 染色质三维结构研究
- 目的:结合Hi-C等技术,研究染色质的三维结构与远距离调控机制。
- 靶标:染色质环中的结合蛋白或增强子。
- 应用:
- 揭示基因组中远程调控元件(如增强子与启动子)的交互作用。
- 示例:
结合ChIP-seq和Hi-C研究增强子与启动子之间的染色质环,分析它们对基因调控的影响。
总结
- ChIP-seq是研究基因组调控的核心工具,应用范围非常广泛。
- 它既能用于揭示蛋白质与DNA结合的基本机制,也能通过结合其他表观遗传学技术,探索基因调控与疾病的深层关系。
- ChIP-seq还在药物研发中起到重要作用,为新靶点的发现提供了重要手段。
部分2:ChIP-seq的实验流程。
ChIP-seq的基本原理与实验流程
ChIP-seq的核心原理是利用抗体特异性捕获与DNA结合的目标蛋白质(如转录因子或组蛋白修饰),然后对这些蛋白结合的DNA片段进行测序和分析,定位蛋白质在基因组中的结合位点。
实验流程:从样本到数据分析
1. 样本固定(Crosslinking DNA-Protein Complex)
- 目的:固定DNA与其结合的蛋白质,保持其相互作用。
- 方法:通常使用甲醛(formaldehyde)将DNA与结合的蛋白质交联,形成稳定的化学共价键。
- 注意事项:
- 固定时间过长可能导致非特异性交联。
- 过短会导致结合蛋白的脱落。
2. 染色质剪切(Chromatin Shearing)
- 目的:将基因组DNA切割成小片段(一般为200-500 bp)。
- 方法:
- 超声波:通过超声波产生物理剪切。
- 酶切:使用特定核酸酶切割DNA。
- 优化重点:
- DNA片段大小的均一性非常重要,通常在实验中会对剪切效率进行检测。
3. 免疫共沉淀(Chromatin Immunoprecipitation, ChIP)
- 目的:利用特异性抗体富集与目标蛋白结合的DNA片段。
- 步骤:
- 添加抗体,抗体特异性结合目标蛋白(如转录因子或修饰的组蛋白)。
- 通过磁珠或蛋白A/G树脂捕获抗体-蛋白-DNA复合物。
- 洗涤去除非特异性结合。
- 关键点:
- 抗体的特异性是成功的关键。
- 磁珠的使用可以显著提高富集效率。
4. 逆交联和DNA纯化
- 目的:分离DNA和蛋白质,并提取纯化DNA。
- 方法:
- 通过加热或化学试剂(如蛋白酶K)解除蛋白质和DNA的交联。
- 用常规的DNA提取试剂盒或苯酚-氯仿方法提取DNA。
- 结果:得到目标蛋白质结合的纯化DNA片段。
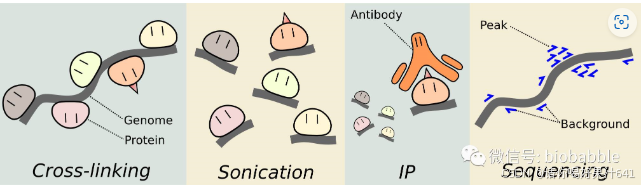
5. 文库构建(Library Preparation)
- 目的:将提取的DNA片段转化为高通量测序平台可用的测序文库。
- 步骤:
- 末端修饰:将DNA片段的末端修复成平端。
- 加接头:添加测序适配器,用于后续测序。
- PCR扩增:扩增DNA片段,增加测序量。
- 注意事项:
- 扩增次数过多可能引入偏倚,需进行优化。
6. 高通量测序
- 测序平台:常用Illumina测序平台(如HiSeq、NovaSeq)。
- 结果:
- 得到目标蛋白结合DNA片段的序列数据(fastq格式)。
数据分析流程
1. 数据质量控制
- 工具:FastQC
- 内容:
- 检测测序数据质量。
- 去除低质量reads和接头序列。
2. 序列比对(Mapping)
- 工具:Bowtie2 或 BWA
- 目的:将测序reads比对到参考基因组(如人类基因组hg38)。
- 结果:
- 生成BAM文件,记录每条reads在基因组中的比对位点。
3. 峰值识别(Peak Calling)
- 工具:MACS2(最常用)现在已经出3了
- 目的:识别基因组中蛋白质结合富集的区域(峰值)。
- 方法:
- 输入对照样本校正背景噪音,生成高置信度的峰值。
- 结果:
- 生成BED文件,记录峰值的位置信息。
4. 峰值注释(Peak Annotation)
- 工具:Homer 或 ChIPseeker
- 目的:
- 将峰值注释到基因组功能区域(如启动子、增强子)。
- 关联结合位点与目标基因。
- 结果:生成结合位点与基因注释的结果。
5. 功能富集分析
- 工具:DAVID、GO、KEGG
- 目的:分析结合的目标基因是否富集在特定的生物学通路中。
6. 可视化分析
- 工具:IGV(基因组浏览器)、deepTools
- 目的:展示目标蛋白结合位点在基因组中的分布及信号强度。
ChIP-seq的关键控制点
- 抗体质量:抗体的特异性和敏感性是整个实验的关键。
- 染色质剪切:剪切效率直接影响后续DNA片段的均一性和信号强度。
- 测序深度:
- 针对窄峰(如转录因子):10-20百万reads。
- 针对宽峰(如组蛋白修饰):40-50百万reads。
- 对照样本:通常需要输入对照样本校正背景噪音。
部分3:ChIP-seq的应用实例与技术优化。
ChIP-seq的主要应用实例
ChIP-seq技术广泛用于解析基因组水平的蛋白质-DNA相互作用和表观遗传修饰模式,其具体应用包括:
1. 转录因子结合位点研究
- 目的:了解特定转录因子在基因组中的结合位点及调控网络。
- 实例:
- p53:通过ChIP-seq确定p53的结合位点,研究其在肿瘤抑制中的调控机制。
- Oct4/Sox2/Nanog:研究这些转录因子在胚胎干细胞中的结合位点,解析其维持干性和分化的调控网络。
- 数据分析:
- 峰值注释到启动子或增强子区域。
- 功能富集分析预测其调控的目标基因。
2. 组蛋白修饰研究
- 目的:探索组蛋白化学修饰(如H3K27me3、H3K4me3)在基因组中的分布,解析表观遗传调控机制。
- 实例:
- H3K4me3:标记启动子区域的活性基因。
- H3K27me3:研究基因沉默相关修饰的基因组分布。
- 特色:
- 修饰类型决定峰的宽度:
- 窄峰(如H3K4me3):信号集中在启动子区域。
- 宽峰(如H3K27me3):信号覆盖整个基因体区域。
- 修饰类型决定峰的宽度:
3. 染色质开放性与结构研究
- 目的:研究染色质状态(如开放或压缩)对基因表达的影响。
- 实例:
- 使用染色质重塑因子(如BRG1)的ChIP-seq分析,探索其如何调控染色质开放,激活基因表达。
- Hi-C结合分析:揭示染色质三维结构如何影响远距离基因调控。
- 技术特色:
- 常结合ATAC-seq或Hi-C数据,进行染色质可及性和三维基因组的联合分析。
4. RNA聚合酶II结合位点研究
- 目的:分析RNA Pol II在基因组中的分布,了解转录起始及转录延伸机制。
- 实例:
- 分析RNA Pol II在不同细胞类型的结合模式,预测哪些基因正在被活跃转录。
- 数据分析:
- 峰值通常与启动子结合。
- 结合RNA-seq数据,评估转录活性的变化。
5. 增强子活性分析
- 目的:鉴定活跃的增强子元件,分析其调控的基因表达。
- 实例:
- 检测H3K27ac(活跃增强子标记)或特定转录因子结合的区域。
- 超级增强子(Super Enhancers):识别广泛分布的增强子富集区域,研究其在细胞命运决定中的作用。
- 工具:
- ROSE(识别超级增强子)。
- Homer(注释增强子附近的目标基因)。
ChIP-seq技术优化
1. 抗体优化
- 问题:抗体特异性和亲和力不足可能导致高背景或信号丢失。
- 优化方法:
- 选择经过验证的商用抗体。
- 进行Western blot检测抗体的特异性。
- 使用多个抗体验证关键结果。
2. 染色质剪切
- 问题:剪切不足会导致低分辨率,过度剪切可能破坏结合位点。
- 优化方法:
- 超声波剪切的强度和时间应预实验优化。
- 常检测200-500 bp的片段分布。
3. 对照样本设计
- 输入对照(Input control):
- 反映背景信号。
- 用于峰值识别时校正背景噪音。
- IgG对照:
- 检测非特异性抗体结合。
4. 测序深度
- 窄峰(如转录因子):
- 推荐10-20百万reads。
- 宽峰(如组蛋白修饰):
- 推荐40-50百万reads。
5. 数据分析工具选择
- 峰值识别:
- MACS2:标准化工具,支持输入对照。
- SICER:适合宽峰标记。
- 峰值注释:
- Homer:注释到基因功能区域。
- ChIPseeker:R语言工具,生成高质量图表。
6. 生物学重复
- 作用:
- 确保实验结果的可靠性。
- 增加统计分析的精度。
- 推荐:
- 至少进行2-3次重复实验。
案例分析:H3K27ac在癌症中的应用
- 研究目标:利用H3K27ac标记的ChIP-seq数据,识别肿瘤细胞中的活跃增强子。
- 分析方法:
- 比较癌细胞与正常细胞的H3K27ac峰值分布。
- 注释超级增强子,预测关键调控基因。
- 研究发现:
- 癌细胞中超级增强子激活了肿瘤驱动基因(如MYC)。
- 应用:
- 提供肿瘤治疗的新靶点。
部分4:ChIP-seq数据质量控制与常见问题。
ChIP-seq数据质量控制
数据质量直接影响ChIP-seq的分析结果,常见的质量控制步骤和关键指标如下:
1. 原始数据质量检查
- 工具:FastQC、MultiQC
- 检查点:
- 碱基质量分布:确保测序读段的碱基质量在Q30以上。
- GC含量分布:与参考基因组一致(人类基因组通常为40%-60%)。
- 接头污染:检查是否存在测序接头序列,并清除污染。
2. 数据比对质量
- 工具:Bowtie2、BWA
- 检查点:
- 比对率:
- 对参考基因组的比对率应达到80%以上。
- 如果比对率过低,可能由于:
- 样本DNA污染。
- 错误的参考基因组版本。
- 重复率:
- 高重复率表明测序深度不足或PCR扩增过度。
- 比对率:
3. 数据冗余过滤
- 目标:去除重复的测序读段以减少假阳性。
- 工具:Samtools、Picard
- 标准:
- 唯一比对的非冗余读段比例应>80%。
- 如果比例较低,可能是实验过程中的DNA量不足或扩增效率不佳。
4. 峰值识别质量
- 工具:MACS2、SICER
- 关键指标:
- 峰值的强度与背景的信噪比。
- 窄峰标记(如转录因子):峰值应清晰集中。
- 宽峰标记(如H3K27me3):峰值应均匀分布。
5. 数据深度和信噪比
- 工具:phantompeakqualtools、deepTools
- 检查点:
- Normalized Strand Cross-correlation (NSC):
- 测量信号与噪声的比例。
- 窄峰标记:NSC > 1.5。
- 宽峰标记:NSC > 1.1。
- 读数深度:
- 窄峰标记:10-20M。
- 宽峰标记:40-50M。
- Normalized Strand Cross-correlation (NSC):
常见问题与解决方案
1. 低信噪比
- 问题:
- 峰值不明显,信号与背景无法区分。
- 原因:
- 抗体特异性差或样品质量问题。
- 染色质剪切不均匀。
- 解决方案:
- 更换高质量抗体。
- 优化染色质剪切条件(如超声波功率和时间)。
2. 高重复率
- 问题:
- 数据中重复读段比例过高。
- 原因:
- 样本起始量不足。
- PCR扩增循环过多。
- 解决方案:
- 提高起始DNA量。
- 减少PCR扩增循环。
3. 比对率低
- 问题:
- 比对到参考基因组的读段比例较低。
- 原因:
- 样品污染或非目标物种DNA。
- 使用了不匹配的参考基因组版本。
- 解决方案:
- 检查样品来源。
- 确保参考基因组版本与实验设计一致。
4. 峰值分布异常
- 问题:
- 峰值异常集中于重复序列区域。
- 原因:
- 样品中重复序列比例高。
- 数据过滤不充分。
- 解决方案:
- 使用PeakRanger或RepeatMasker进行重复区域校正。
- 调整峰值识别参数,增加背景校正。
5. 数据中存在批次效应
- 问题:
- 不同实验之间数据不可比。
- 原因:
- 不同实验条件或测序平台。
- 解决方案:
- 采用分位数归一化(Quantile Normalization)。
- 在实验设计中统一测序批次。
6. 样本重复间一致性差
- 问题:
- 生物学重复的峰值或信号分布差异大。
- 原因:
- 样本质量不均一。
- 操作流程不一致。
- 解决方案:
- 增加生物学重复(至少3次)。
- 标准化实验流程。
质量评估的关键工具与指标
| 工具 | 功能 | 关键指标 |
|---|---|---|
| FastQC | 原始数据质量评估 | Q30、GC含量 |
| Bowtie2/BWA | 比对到参考基因组 | 比对率、重复率 |
| MACS2/SICER | 峰值识别 | 峰值强度、背景校正 |
| phantompeakqualtools | 信噪比评估 | NSC、RSC |
| deepTools | 数据可视化与信号评估 | 热图、Profile图 |
| ChIPQC | R包,全面评估数据质量 | 样本间一致性 |
部分5:ChIP-seq高级数据分析方法与整合策略。
高级数据分析方法
ChIP-seq的高级分析旨在从峰值数据中提取生物学意义,结合其他组学数据,揭示调控机制和功能。
1. 峰值注释(Peak Annotation)
- 目的:确定峰值位于基因组的功能区域(启动子、增强子、内含子等)。
- 工具:
- Homer:
- 提供详细的基因注释。
- 输出基因区域分布图。
- ChIPseeker:
- R语言工具,生成峰值与基因区域的热图。
- Homer:
- 应用:
- 鉴定调控元件(启动子、增强子)。
- 确定与转录因子或修饰相关的基因。
2. 差异结合分析(Differential Binding Analysis,DBA)
- 目的:识别不同条件下结合强度显著变化的峰值。
- 工具:
- DiffBind:
- 基于DESeq2,分析差异结合位点。
- csaw:
- 使用滑动窗口方法分析差异。
- DiffBind:
- 应用:
- 比较处理组与对照组的结合位点差异。
- 探索药物或环境因子对结合模式的影响。
3. 基序分析(Motif Analysis)
- 目的:识别峰值区域的转录因子结合位点。
- 工具:
- Homer:
- 定位已知和新的Motif。
- MEME:
- 灵活的Motif发现工具。
- Homer:
- 应用:
- 预测调控因子。
- 鉴定潜在的新结合模式。
4. 功能富集分析(Functional Enrichment Analysis)
- 目的:分析目标基因是否富集在特定生物学过程或信号通路中。
- 工具:
- DAVID:提供基因功能注释。
- ClusterProfiler:R语言工具,支持GO和KEGG富集分析。
- 应用:
- 解析峰值基因的功能角色。
- 识别疾病相关通路。
5. 信号强度归一化与可视化
- 目的:直观展示峰值信号及其差异。
- 工具:
- deepTools:
- 绘制信号热图和Profile图。
- IGV:
- 在基因组范围内可视化信号。
- deepTools:
- 应用:
- 比较不同样本的信号分布。
- 验证显著峰值的可靠性。
6. 多组学数据整合
- 目的:将ChIP-seq与RNA-seq、ATAC-seq等数据联合分析。
- 策略:
- ChIP-seq + RNA-seq:
- 分析结合位点是否影响基因表达。
- ChIP-seq + ATAC-seq:
- 结合染色质开放状态鉴定活跃结合位点。
- ChIP-seq + RNA-seq:
- 应用:
- 构建转录因子调控网络。
- 探索表观遗传调控与转录的关系。
整合策略与功能验证
1. 构建调控网络
- 目的:揭示转录因子如何通过结合元件调控基因表达。
- 方法:
- 结合ChIP-seq Motif分析和RNA-seq差异基因。
- 构建调控因子与靶基因的网络。
2. 验证关键结合位点
- 实验验证:
- qPCR:验证关键结合位点的富集情况。
- CRISPR/Cas9:删除关键结合元件,观察基因表达变化。
- 工具:
- IGV可视化峰值。
- Primer设计工具设计验证引物。
3. 跨物种分析与进化研究
- 目的:研究调控机制的保守性。
- 方法:
- 比较不同物种中ChIP-seq数据。
- 分析保守Motif和功能元件。
数据整合案例
案例:转录因子P53的癌症调控研究
- ChIP-seq数据:定位P53的结合位点。
- RNA-seq数据:分析P53靶基因的表达变化。
- 功能富集:
- GO:富集在细胞周期调控、DNA修复。
- KEGG:P53信号通路显著。
- 验证:
- qPCR验证关键结合位点。
- CRISPR干扰增强子,观察细胞周期基因表达变化。
部分6:ChIP-seq结果解读与报告撰写。
ChIP-seq结果解读的核心要素
1. 峰值分布的全面分析
- 分布概览:
- 统计峰值在基因组功能区域的分布(启动子、增强子、外显子等)。
- 可视化不同区域中峰值的百分比。
- 关键问题:
- 峰值主要分布在基因启动子附近还是远程增强子区域?
- 这些峰值与哪些生物学功能相关?
2. 差异结合的显著性
- 核心结果:
- 在不同条件或样本间识别显著的结合位点差异。
- 用热图或火山图展示差异结合的峰值。
- 解读:
- 哪些基因的转录因子结合发生了显著变化?
- 这些变化可能与哪种调控机制或病理过程相关?
3. 目标基因功能的解析
- 功能富集分析:
- 通过GO和KEGG通路注释目标基因。
- 实例:
- 某转录因子结合的目标基因富集在“细胞周期调控”和“DNA损伤修复”路径上,可能暗示其参与细胞增殖异常。
4. 结合位点的保守性
- 跨物种分析:
- 比较结合位点在不同物种中的保守性。
- 实例:
- P53的结合位点在人类和小鼠中高度一致,提示其核心调控作用具有进化保守性。
5. 数据整合与模型构建
- 整合多组学数据:
- 将ChIP-seq与RNA-seq结果结合,分析结合位点是否影响基因表达。
- 模型示例:
- 构建转录因子调控网络,明确结合位点对目标基因表达的正/负调控作用。
撰写报告的逻辑结构
1. 背景介绍
- 研究问题:明确转录因子或组蛋白修饰的生物学意义。
- 方法概述:简单描述ChIP-seq的实验设计。
2. 数据与方法
- 数据处理流程:
- 原始数据质量控制、比对、峰值识别和注释等。
- 使用软件和参数:
- 明确工具(如MACS2、Homer)及其参数设置。
3. 主要结果
- 峰值分布:
- 图表展示不同区域峰值的分布(如启动子、增强子)。
- 功能分析:
- 描述目标基因的功能富集结果。
- 突出重要通路或调控网络。
- 差异结合分析:
- 强调显著结合差异及其潜在生物学意义。
4. 讨论与结论
- 结果总结:
- 结合结果解释转录因子或组蛋白修饰的作用。
- 关联生物学问题:
- 讨论结果与疾病、发育或其他机制的相关性。
- 展望与局限:
- 展望后续研究方向。
- 提出可能影响结果的技术或分析限制。
关键可视化图表
- 质量控制图:
- FastQC生成的碱基质量图。
- 峰值分布图:
- 不同功能区域的峰值分布饼图或柱状图。
- 差异结合热图:
- 展示不同样本的结合强度差异。
- 功能富集柱状图:
- 富集通路的显著性统计图。
- 调控网络图:
- 描绘转录因子与目标基因的调控关系。
实例:ChIP-seq研究P53的调控作用
-
研究问题:
- P53在基因组中的结合位点与其调控的基因网络如何影响细胞周期和肿瘤抑制?
-
主要发现:
- 峰值主要分布在启动子区域(>60%)。
- GO分析显示目标基因显著富集于“细胞周期停滞”和“DNA修复”路径。
- RNA-seq数据显示,与P53结合位点相关的基因表达上调显著。
-
结论:
- P53通过直接结合调控细胞周期关键基因(如CDKN1A)实现抑癌作用。
- 其结合位点的分布模式在癌细胞和正常细胞中存在显著差异。
下一步行动
如果您对报告结构清楚了,可以继续深挖 具体分析的高级问题 或 如何优化ChIP-seq实验设计。

















 6685
6685

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









