







https://onlinelibrary.wiley.com/doi/10.1111/tpj.14193
https://github.com/Pang-Junling/maize-5DAP-eQTL
摘要
在玉米中,籽粒性状强烈影响整体谷物产量,众所周知,基因表达的复杂时空程序会协调籽粒发育,因此提高我们对籽粒发育的了解有助于提高籽粒产量。在这里,使用授粉后 5 天和 15 天 (DAP) 玉米粒的表型、基因型和转录组学数据进行大型关联绘图组,我们采用了多种定量遗传学方法——全基因组关联研究 (GWAS) 以及表达数量性状基因座 (eQTL) 和数量性状转录本 (QTT) 分析——以深入了解玉米籽粒发育的分子遗传基础。这导致在 5 DAP 处鉴定了 137 个推定的内核长度相关基因,其中 43 个位于先前报道的 QTL 区域。引人注目的是,我们鉴定出了一个 eQTL,它与编码最近描述的 m 的玉米同源物的基因座重叠6来自拟南芥的甲基化读取蛋白 ECT2;这个假定的EPI 系列eQTL 与 53 个基因相关,可能代表内核发育的主要表观转录组调节因子。值得注意的是,在与此相关的基因中EPI 系列eQTL,10 个用于玉米胚乳中的主要储存蛋白 (zeins),两个是玉米醇溶蛋白表达或胚乳发育的已知调节因子 (Opaque2 和 ZmICE1)。总的来说,除了对与玉米籽粒发育相关的大量 eQTL 的基因组属性进行编目和表征外,我们的研究还强调了 eQTL 方法如何增强 GWAS 和 QTT 研究的影响,并推动对植物基础生物学的见解。
介绍
玉米 (Zea mays L.) 不仅作为食品、饲料和工业产品来源具有世界重要性,而且还是一种具有长期影响力的生物模式物种,表现出广泛的表型和遗传多样性(Hake 和 Ross-Ibarra,2015 年)。 与其他种子一样,它的内核是为植物生长和发育提供重要组成部分的主要储存器官(Scanlon 和 Takacs,2009 年)。收获的籽粒数量及其个体籽粒大小——两个对产量产生巨大影响的复杂遗传性状——是籽粒发育的多步骤和严格调节过程的结果(Li et al., 2013b;Chen et al., 2014;Doll et al., 2017)。因此,推进我们对籽粒发育背后的遗传甚至分子遗传学机制的了解,将对正在进行的全球寻求优化玉米籽粒产量的努力产生重大影响。
关联图谱,也称为连锁不平衡 (LD) 图谱,利用自然种群中的重组事件来利用遗传多样性信息,有可能将复杂性状的表型变异解析到单个基因甚至单个核苷酸的水平。与人类的全基因组关联研究 (GWAS) 不同,农作物中的 GWAS 通常基于通常称为关联面板的永久资源——一个由多样化(最好是纯合子)遗传系组成的群体,只需要进行一次基因分型,但可以针对许多性状进行重新表型分析。玉米,部分由于其杂交性质和随之而来的快速 LD 衰变(在 ∼2 kb 以内),以及其巨大的遗传多样性,是强大的 GWAS 方法持续发展的重要模型(Huang 和 Han,2014)。然而,使用 GWAS 的研究人员必须考虑和管理已知的种群结构在驱动假阴性率增加和假阳性率降低方面的巨大影响。事实上,即使经过仔细管理,仍然不会在关联面板的 GWAS 中检测到一些真正有影响力的基因座(假阴性),因为数量性状位点 (QTL) 等位基因分布与种群结构相关(Bazakos等人,2017 年)。
一种有助于扩大大型定量遗传学研究范围的有趣概念方法称为表达数量性状位点 (eQTL) 分析。这种越来越流行的方法基于对基因表达水平和遗传变异进行关联分析,它可以识别影响基因表达水平的遗传多态性;在构建基因调控网络时,它也有助于在调节因子和靶标之间建立联系(Hansen等人,2008 年;Kliebenstein, 2009;Hammond et al., 2011)。此外,eQTL 信号还可以揭示 GWAS 研究中的本地和远处性状相关变异,从而将表达数据和 DNA 序列变异与表型联系起来并解决复杂性状(Potokina等人,2008 年;Li et al., 2013b;Albert 和 Kruglyak,2015 年)。重要的是,还表明 eQTL 与**数量性状转录本 (QTT) **分析的整合可以促进复杂性状的致病基因的鉴定(Passador-Gurgel et al., 2007;Petretto et al., 2008)。
现在认识到,一系列最终与产量相关的关键发育过程(例如 合胞体形成、细胞化、细胞分化等)发生在玉米籽粒发育的早期 [从授粉到授粉后约 15 天 (DAP);Sabelli 和 Larkins,2009 年]。众所周知,这一发展阶段受基因表达的复杂时空程序的调节(Sekhon等人,2011 年;Li et al., 2014;Doll et al., 2017)。在这里,使用玉米籽粒在 5 DAP 和 15 DAP 的表型、基因型和转录组学数据,为由热带、亚热带和温带自交系组成的大型关联绘图面板,我们进行了 eQTL 、 QTT 和 GWAS 分析,并整合了结果,以深入了解玉米籽粒发育的遗传和分子遗传基础。我们注意到了与发育阶段特异性 eQTL 相关的几个有趣趋势,以及这些调控区域与其相关遗传位点的距离。我们还发现了一个 eQTL,它与编码最近描述的 m 的玉米同源物的基因座重叠6来自拟南芥的甲基化读取蛋白 ECT2;这个假定的EPI 系列eQTL 与 53 个基因相关,显然调节玉米粒中初级蛋白质储存蛋白的代谢。除了强调 eQTL 分析在识别复杂性状的多级基因调控机制方面的效用之外,我们还确定了一种假定的EPI 系列与玉米 ECT2 位点重叠的籽粒性状的 eQTL 说明了定量遗传学方法如何导致基本的生物学见解。
结果与讨论
玉米关联面板的基因分型和 5 DAP 时籽粒中表达基因的表达数量性状位点分析
来自 5-DAP 籽粒 RNA-seq 数据的独特映射读数用于 282 个玉米自交系中每个品系的单核苷酸多态性 (SNP) 检出和基因分型(表 S1)。经过质量控制和插补,我们共鉴定出 663 810 个高质量 SNP。当我们将这些 5-DAP SNP 与之前报道的 15 DAP 玉米粒的 SNP 进行比较时(Fu et al., 2013),我们发现 308 058 个 SNP(5 DAP 的 46.41% 和 15 DAP 的 55.15%)在两个数据集中是共同的(图 1a)。请注意,来自 15 个 DAP 的 SNP 来自两个分析来源连接:使用 MaizeSNP50 BeadChip 确定 49 256 个(Li等人,2012 年);509 322 是从 RNA-seq 读数中调用的。作为另一种 SNP 质量控制措施,我们比较了 5 和 15-DAP SNP 的一致率,发现 5-DAP SNP 与 BeadChip 和 RNA-seq 衍生的 15-DAP SNP 分别具有 97.26% 和 94.71% 的一致性(图 1b)。
在进行 eQTL 分析之前,为了提高作图能力,我们通过结合 5-DAP 和 15-DAP SNP 数据集来扩大 SNP 密度——这导致总共 914 330 个 SNP 覆盖 28 280 个基因,比之前报道的 15-DAP 内核 SNP 数据中涵盖的基因多 3 105 个基因。在本研究中使用的 SNP 中,89.73% 位于基因区域(图 S1a),考虑到 SNP 是从为大型关联群体的每个品系收集的基因表达数据中鉴定的,这是一个合理的分布。每个基因的平均 SNP 数为 32 (图 S1b)。为了准备用于关联分析的数据集,我们检查了所有覆盖基因的表达水平,并消除了种群中位表达水平为零的基因,从而保留了 27 103 个在 5 DAP 下表达的覆盖基因。考虑到玉米粒是一个复杂的结构,并且一个自交系中的时间形态分期不一定与另一个自交系中的相同分期相关,我们探讨了自交系在 5 DAP 时的发育一致性。按自交系中的平均表达量对基因进行排序,平均表达量与每个样品前 1 000 个基因表达量的相关性用作一致性指标。在所有 282 个自交系中,185 个 (65.6%) 显示出非常强的相关性 (r ≥ 0.8) 和 86 个 (30.5%) 强相关性 (0.6 ≤ r < 0.8;Evans, 1996),表明大多数品系都很好地放在一起(表 S2)。在接下来的 eQTL 研究中,相关性不佳的左侧自交系不应产生假阳性,但可能会降低检测能力。
使用混合线性模型 (MLM) 进行关联分析,使用这 27 103 个基因中每个自交系在 5 DAP 和 776 254 个次要等位基因频率 (MAF) 大于 0.05 的整合 SNP 的表达水平数据。然后使用 Benjamini-Hochberg (BH) 方法选择显著相关的 SNP,错误发现率 (FDR) 低于 0.05 (P < 2.6E-6)。随后基于关联分析中的重要 SNP 定义 eQTL 区域使用了先前描述的方法(Fu et al., 2013)。简而言之,当至少 3 个重要的 SNP 聚集在一个基因组位点上,并且没有相距超过 5 kb 的 SNP 对时,这些 SNP 被视为候选 eQTL。在单个基因与多个候选 eQTL 相关的情况下,并且两个候选 eQTL 具有 LD (r2> 0.1),然后去除显著性较低的 eQTL;如果两个候选 eQTLs 具有相同的 eQTL 结合强度 (P 值),则保留具有较大联合效应的 eQTL。
总共鉴定了 22 966 个 eQTL,与 5-DAP 玉米粒中 18 377 个基因的表达相关(图 1c;表 S3)。这些 eQTL 的平均长度为 4 086 bp,平均由 13 个 SNP 组成。回想一下这些主要来自 RNA-seq 分析,大多数 5-DAP eQTL (70%) 与基因区域 (外显子 37.4% 和内含子 32.6%) 重叠也就不足为奇了。根据先前发表的玉米 eQTL 分析,使用 20 kb 作为临界值来定义“局部 eQTL”与“远距离 eQTL”,我们还将 eQTL 分为两种类型(Fu et al., 2013):11 858 (51.6%) 是局部 eQTL,11 108 (48.4%) 是远距离 eQTL。以基因 ZAG2 (GRMZM2G160687) 为例,局部 eQTL 区域由基因座内的 25 个显著相关的 SNP 定义(图 1d)。基于针对该基因的候选 eQTL 区域中最强相关的 SNP 的单倍型分析在玉米关联面板群体中定义了两个单倍型,并且有两种不同的 ZAG2 表达模式表征了这些单独的单倍型(图 1e,f),从而说明了 eQTL 分析在关联面板的自交系中发挥作用的活性调节序列的能力。
通过整合表达数量性状位点、数量性状转录本和全基因组关联研究分析来鉴定早期内核发育相关基因
接下来,我们进行了 QTT 分析,以探索在 5-DAP 内核中表达的与内核长度相关的基因。该分析采用线性回归模型来表征关联面板中每个自交系已发表的内核长度数据之间的关系 (Liu et al., 2016) 与种群中每个自交系中与 eQTL 相关的 18 377 个基因中每个基因的表达数据之间的关系。使用 P < 0.05 的临界值和群体中位数>表达水平 1,905 个与 eQTL 相关的基因被认为是显着的 QTT。另外,我们使用每个自交系的内核长度表型数据和上述 776 254 个 SNP 进行了 MLM 和 MLM。与玉米种群中基于 MLM 的复杂性状关联分析一样,即使在松散的临界值 (1.3E-6;图 2a、上图和 S2;Yang et al., 2014)。因此,我们使用了另一种选择——一般线性模型 (GLM)——进行关联分析,揭示了 12 159 个显著相关的 SNP,涵盖 3 627 个基因(图 2a,底部)。然而,由于众所周知基于 GLM 的关联研究具有很高的假阳性率,尤其是在种群结构不受控制的情况下(Larsson等人,2013 年),我们只保留了 GLM 和显着 QTT 结果共有的基因(图 2b)。因此,我们鉴定了 137 个推定与 5 DAP 的内核长度相关的基因(表 S4)。值得注意的是,这些基因中有 43 个 (31.39%) 位于先前报道的内核长度的 QTL 区域(表 1)。
对我们的综合 eQTL、QTT 和 GWAS 分析中鉴定的 137 个基因的基因本体论 (GO) 分析表明,细胞周期相关过程在我们推定的内核长度相关基因中富集 (P = 2.0E-4;图 S3)。排名第一的内核相关转录本(来自 QTT 分析)为 GRMZM2G099074;最接近的同源物(蛋白质序列的 55% 同一性;E = 3.0E-118) 是胚胎缺陷 1379 (AtEMB1379 或 AtNSE1,AT5G21140),该基因在调节 G2/M 阶段的细胞周期和内复制中具有既定功能 (Liet al., 2017)。GRMZM2G099074表达与籽粒长度之间的相关性很高 (Spearman ρ = 0.38,P < 3.5E-12; 图 2c)。此外,基于GRMZM2G099074 eQTL 区域中最强相关性 SNP 的单倍型分析定义了两种单倍型,并且这两种单倍型之间的内核长度存在明显且显着的差异(图 2d)。
我们发现GRMZM2G099074和内核长度之间存在很强的关联,以及细胞周期调控相关基因的过度表达,这表明在内核发育的早期阶段对与细胞周期调控相关的基因表达的严格控制对于确定成熟内核的最终大小可能至关重要。支持这一观点,我们发现与推定编码有丝分裂纺锤体检查点蛋白 MAD2 (GRMZM2G112072) 的基因重叠的 eQTL 与预测的染色质重塑蛋白 (GRMZM2G138125) 的表达相关。已知水稻中 GRMZM2G138125 的同源物对于胚乳发育早期的合胞体发育至关重要(Hara et al., 2015),强烈表明该基因在玉米粒发育中具有类似的作用。
5 个 DAP 和 15 个 DAP 的表达数量性状位点比较
接下来,我们将 5 个 DAP 的 eQTL 与 15 个 DAP 的 eQTL 进行了比较。为了最大限度地提高两个 eQTL 数据集之间的可比性,通过保留任何相交 eQTL 的最大基因组区域,合并从 5 个 DAP 和 15 个 DAP 中鉴定的所有 eQTL 区域(图 3a)。因此,我们获得了 16 327 个合并的 eQTL 区域,平均长度为 4 418 bp,平均由 35 个 SNP 组成,分别与 5-DAP 和 15-DAP 内核的 18 377 和 18 872 个基因相关(表 S5)。与 SNP 的数据来源一致(基于 RNA 表达),大多数合并的 eQTL (71%) 与基因区域共享重叠(39% 在外显子中,32% 在内含子中),并且这些 eQTL 中的 22% 与 UTR 序列共享重叠 [此处定义为转录起始位点 (TSS) 或终止位点的 1 kb 以内];其余 7% 的 eQTL 落入基因间区域 (图 3b)。
总共在两个阶段的发育内核中普遍检测到 9 106 个 eQTL (占合并 eQTL 数据集的 55.8%) (‘stage-sharing’ eQTLs)。还有 3 531 个 ‘5 个 DAP 特异性’ eQTL 和 3 690 个 ‘15 个 DAP 特异性’ eQTL (图 3c)。仔细观察阶段共享与阶段特异性 eQTL 的物理分布揭示了一个有趣的趋势:对于具有相关 eQTL 的给定基因,阶段特异性 eQTL 到相关基因 TSS 的距离大于阶段共享 eQTL(图 3d)。这里的一个可能含义是,在相对较窄的时间/发育窗口内发挥其调节功能的 eQTL 可能相对更有可能使用远距离机制。检查其他植物基因组中是否存在类似趋势将很有趣,因为它们经历了不同的发育序列。与 5-DAP 特异性 eQTL 相关的基因的 GO 功能类别分析富集了植物对刺激的反应,而 15-DAP 特异性 eQTL 的靶标富集了养分库活性,这一发现与已知的 15 DAP 时谷物填充的生理学一致。阶段共享 eQTL 的靶标在发育相关过程中富集,如多细胞生物发育、解剖结构发育和细胞器组织(图 S4)。
对 5 DAP 与 15 DAP 时阶段共享 eQTL 对单个靶基因的调控作用的分析表明,9 106 个阶段共享 eQTL 中有 324 个表现出“反向调节活性”(图 3e)。例如,具有两个等位基因(‘CC’ 或 ‘GG’)的 eQTL 在 5 DAP 和 15 DAP 时与 GRMZM2G059580(编码延伸因子 1-γ 的基因)的表达显著相关,但每个特定等位基因的该基因的表达在两个发育阶段之间是倒置的:带有 ‘CC’ 等位基因的品系在 5 DAP 时 GRMZM2G059580 表达相对较低, 而具有相同等位基因的品系在 15 DAP 时具有相对较高的 GRMZM2G059580 表达(图 3f)。在人类的 eQTL 研究中,对于单个等位基因的组织特异性反向调节作用,也报道了类似的现象(Fu et al., 2012)。值得注意的是,这种 eQTLs 在人类中的比例与我们在玉米粒发育中观察到的相似。当我们仔细检查这些 eQTL 与其相关基因的距离时,我们发现 324 个这样的 eQTL 中有 323 个是局部 eQTL(表 S6)。请注意,相对于整组阶段共享的 eQTL 的分布模式,这被过度代表了,其中 51.1% 的 eQTL 是局部的(卡方检验,P < 2.2E-16)。在 5 DAP 和 15 DAP 时观察到具有反向效应的 eQTL 可能表明不同发育阶段基因表达的不同调控机制,但我们不能排除在为两个数据集收集样本时,不同生长条件影响的表达变化可能引起反向效应的可能性。
与多个基因和候选基因相关的表达数量性状位点 EPI 系列内核性状的 eQTL
在我们的研究中,大约 90% 的 eQTL 与少于三个靶点相关 (91.4% 在 5 DAP 和 90.1% 在 15 DAP),超过 50% 的 eQTL 仅与一个基因相关。我们将与三个以上靶标相关的 eQTL 定义为“多靶点 eQTL”;在这个阈值下,分别鉴定出 1 083 个和 1 265 个多靶点 eQTL 的 5 个 DAP 和 15 个 DAP(表 S7)。分别有 53 个和 79 个 5 个 DAP 和 15 个 DAP 的阶段特异性多靶点 eQTL(表 S8 和 S9)。GO 分析显示,5-DAP 特异性多靶点 eQTL 的靶基因富集了对非生物刺激的反应以及前体代谢物和能量的产生相关的功能(表 S10),而与 15-DAP 特异性多靶点 eQTL 相关的基因富集了营养库活性(表 S11)。
我们发现了一个明显极其重要的多靶点 eQTL 位于 7 号染色体上 (chr7:8310530…8315224) 在 15 个 DAP 处具有特异性,并与分布在玉米基因组所有 10 条染色体上的 53 个基因相关。该 eQTL 跨越 GRMZM2G144726 位点,该位点位于先前报道的籽粒重量的 QTL 区域内(图 4a;Liu等人,2017 年),并且在仁发育过程中,根据已发表的表达图谱,在胚乳、胚和整个仁中高度表达(图 S5;Chen等人,2014 年)。基于该 eQTL 中最重要的 SNP (S7_8311142) 的单倍型分析表明,在关联面板中,它有两种单倍型。内核表型存在显著差异:两种单倍型中的一种具有更长、更厚的内核,并且具有更高的 100 粒重(“大内核单倍型”;图 4b)。在 53 个相关基因中,10 个是玉米醇溶蛋白的基因,玉米醇溶蛋白是玉米胚乳中最丰富的储存蛋白(图 4c;Thompson 和 Larkins,1994 年)。同样值得注意的是,不透明 2 基因 (O2, GRMZM2G015534) 是玉米醇溶蛋白基因表达的长期研究和知名调节因子 (Zhang et al., 2015),是该 eQTL 的 53 个靶标之一。同样,碱性螺旋-环-螺旋 (bHLH) 基因 ZmICE1 (GRMZM2G173534) — 编码一种与 O11 强烈相互作用的蛋白质,O11 是胚乳发育和营养代谢的中心调节因子,直接调节上游 O2 的表达 (Feng et al., 2018)——也在 53 个相关基因中。除此之外,ZmICE1 、 O 2 以及 10 个玉米醇溶蛋白基因在大核单倍型中的表达水平显著升高(图 S6)。
令人着迷的是,该 eQTL 覆盖的 GRMZM2G144726 位点编码拟南芥 ECT2 基因 (AT3G13460) 的同源物,该基因编码一种最近表征的表观转录组标记 N6-甲基腺苷 (m6一个;Arribas-Hernández等人,2018 年;Scutenaire et al., 2018;Wei et al., 2018;图 4d,e)。对于上下文,在概念上类似于 DNA 甲基化的过程中,m6mRNA 上的 A 标记可以动态写入、擦除和读取,已知这些标记会影响基因调控。ECT2 是最早报道的 m 之一6A 拟南芥读者;它是正常毛状体所需的 YTH 结构域家族蛋白(Arribas-Hernández等人,2018 年;Scutenaire et al., 2018;Wei等人,2018 年)和叶子发育(Arribas-Hernández等人,2018 年)。据透露,m6拟南芥中的 A 主要出现在 RRACH 基序中(Shen et al., 2016),而最近的一项研究发现 ECT2 可能结合 m6A/RNA 通过拟南芥中的 URUAY 基序 (Wei et al., 2018)。为了追求与 GRMZM2G144726 位点重叠的 eQTL 可能与控制内核发育的某些表观转录组机制有关的想法,我们在 53 个靶基因的转录本中搜索了 RRACH 和 URUAY 基序。我们发现,与 RRACH 基序相比 (背景中 98.1% 对 99.2%,P = 0.29) 相比,53 个基因中转录区域中 URUAY 基序的富集略显著 (背景中为 79.2% 对 69.7%,P = 0.29),尽管这两个基序都以高频率存在。我们没有发现 URUAY 基序 (P = 0.15) 和 RRACH 基序 (P = 0.15;表 S12)。需要进一步的实验来确定 m6在两种单倍型之间显著差异表达的所有 53 个基因中的一个基序(表 S13)。
拟南芥 ECT2 作为 m 的功能6读者取决于 YTH 结构域的存在(Arribas-Hernández等人,2018 年;Scutenaire et al., 2018;Wei et al., 2018)。为了探索 eQTL 区中GRMZM2G144726位点重叠的 SNP 可能改变 YTH 结构域的氨基酸序列从而影响其功能的可能性,我们注释了该 eQTL 区中的所有 48 个 SNP,发现其中只有 3 个位于 GRMZM2G144726 的推定 YTH 结构域中(表 S14)。这三个 SNP 在 LD 下,与 GRMZM2G144726 最显着相关的 SNP (S7_8311142) (图 S7a)。其中两个是同义变体,而另一个是非同义变体,这导致 Glu466 变为 Asp466,Asp466 是 YTH 结构域的非保守氨基酸(图 S7b,c;Arribas-Hernández et al., 2018),这表明这些变体不太可能改变 m6GRMZM2G144726 YTH 结构域的 A/RNA 结合能力。鉴于 GRMZM2G144726 在两种单倍型中显著差异表达(表 S13),我们推测可能存在一种影响 GRMZM2G144726 转录水平的未知机制,以影响其在自交系种群中内核发育中的功能。
这些数据表明,我们的定量遗传学分析已经确定了EPI 系列eQTL 用于内核发育的主要表观转录组调节因子 (GRMZM2G144726,以下简称 ZmECT2)。主要产量性状受到表向转录组调控的可能性是一个令人兴奋的想法,我们对关联面板系中 5-DAP 和 15-DAP 内核的转录组分析可以作为探索这些想法的初始数据集。然而,需要专门监测目标转录本的标记和未标记形式的实验,以指导未来对 m 分布的研究(除其他问题外)6靶转录本上的 A 结合基序、靶标 mRNA 周转速率、标记与未标记靶转录本的细胞定位和群体,以及遗传纵 ZmECT2 对上述所有主题的影响。
实验程序
植物种质、RNA 制备和 Illumina 测序
用于 5-DAP 玉米粒研究的品系总数为 282 株,其中 268 株来自先前 15-DAP eQTL 分析中使用的 368 个自交系(Yang et al., 2010;Fu et al., 2013) 和其他 14 个是新添加的温带自交系。所有 282 个品系均于 2014 年在海南省(中国)以单行样块种植,使用具有三个重复的不完全随机区组设计,这与之前的 15-DAP 设计相同,但 15-DAP 样品的种植地点是 2010 年湖北省荆州市(中国)。每个区块中的 8 个穗是自花授粉的,每个区块的 3-4 个穗中的 10-20 粒种子以 5 DAP 收集。将收集的 3 个复制的种子混合以提取总 RNA。根据产品说明,使用 RNAprep 纯植物试剂盒(TIANGEN,Beijing,China)提取总 RNA。使用 oligo (dT) 积累总 mRNA,并使用制造商指定的方法(Illumina,San Diego,USA)进行 Illumina 方案的文库构建。使用 Illumina HiSeq 2500 测序平台对每个样本进行 125 bp 的双端测序。
读取映射
去除接头和低质量序列后,对测序的读数进行清洗,并保留大于 50 bp 的读数以供进一步分析。使用 TopHat2 (Kim et al., 2013) 将清理后的读数映射到玉米 B73 AGPv3 参考基因组,关键参数如下:‘-i 5 -I 60000 –library-type fr-unstranded -r 250 –mate-std-dev 60 –no-novel-juncs –microexon-search’。使用 Picard 工具 (http://picard.sourceforge.net) 中的 ‘CollectInsertSizeMetrics.jar’ 插件估计配对读数的插入大小 (−r) 和标准差 (–mate-std-dev)。仅保留可以唯一映射到基因组的读数以供进一步分析。
基因表达水平的定量
使用 GenomicFeatures 和 GenomicAlignments 包计算注释基因 (AGPv3, Ensembl release 27) 的读取计数 (Lawrenceet al., 2013)。DESeq2 包 (Love等人,2014) 用于标准化样本之间的读取计数并计算每个基因的 FPKM 值。保留群体中位表达水平> 0 的基因用于关联分析。为了在进行关联分析时避免异常值和基因表达值非正常性的影响,使用 R 中的 qqnorm 函数 (Ihaka 和 Gentleman, 1996) 对每个基因的表达值进行标准化。
单核苷酸多态性检出和插补
GATK(版本 v3.6;McKenna et al., 2010)应用于 SNP 调用。参数是根据 RNA-seq 的 GATK 最佳实践管道设置的(DePristo等人,2011 年;Van der Auwera et al., 2013)。简而言之,通过设置检出变体 20 (-stand_call_conf 20) 的最小 phred 标度置信阈值,然后根据 Fisher 链偏差 < 30.0 和深度质量 > 2.0 过滤,为每个样本单独调用 SNP。然后合并来自不同样本的 SNP,并通过添加参数 ‘-gt_mode GENOTYPE_GIVEN_ALLELES’ 使用 HaplotypeCaller 调用一些基因座的缺失信息。在 SNP 调用过程中,使用 Picard 工具对映射文件中的读长进行重新排序和排序,并标记重复项。已知的玉米插入缺失信息可从 Ensembl Plants 网站 (ftp://ftp.ensemblgenomes.org/pub/plants/release-27/vcf/zea_mays/zea_mays.vcf.gz) 下载。在插补前过滤掉杂合子率> 10% 或缺失率> 60% 的 SNP。记录杂合基因型 (0/1) 并替换为缺失 (./.)。然后计算每个 SNP 的 MAF,MAF < 2% 的 SNP 也被丢弃。
BEAGLE v4.1 (Browning and Browning, 2016) 用于插补缺失基因型,因为它在低覆盖度测序数据的高缺失率下具有良好的性能(Pasaniuc et al., 2012;Swarts et al., 2014)。我们按照 CONVERGE 研究(CONVERGE 联盟,2015 年)中建议的策略应用 BEAGLE,发现相对于其他方法具有更高的插补准确性(Davies et al., 2016)。我们首先在没有参考面板的情况下对所有站点进行估算,然后用参考面板进行估算(Bukowski等人,2018 年)。通过将前者替换为后者(如果存在)来合并两组插补结果。插补后的杂合基因型和在 SNP 检出程序中被记录为杂合位点的插补基因型都被掩盖为缺失 (./.)。
将本研究中调用的基因型与玉米 15-DAP 研究中调用的基因型进行了比较(Fu et al., 2013)。根据来自相同玉米自交系的 SNP 计算一致性。合并两个数据集的 SNP 以进行进一步分析。对于重叠的 SNP 位点,保留了本研究中调用的基因型 (5 DAP)。VariantAnnotation 包 (Obenchainet al., 2014) 用于注释 SNP 并根据其基因组位置将其分为五个区域 (5’-UTR、CDS、Intron、3’-UTR 和 Intergenic 区域)。GenomicFeatures 包 (Lawrenceet al., 2013) 用于读取“gtf”格式文件以进行基因注释。
基因表达谱的种群结构、亲缘关系估计和隐藏因子分析
在关联分析之前,主成分和亲缘关系矩阵是根据 236 205 个 SNP 计算的,这些 SNP 没有缺失,并且使用 TASSEL v5 的 MAF > 0.05(Bradbury等人,2007 年)。前三个主成分用作种群结构,亲缘关系估计使用 ‘Normalized_IBS’ 方法。
使用 PEER (Stegle et al., 2010) 分析基因表达变异的隐藏因素,它使用贝叶斯框架来解释复杂的非遗传因素。在分析过程中,前三个主成分(种群结构)被用作模型的协变量。我们使用了之前的参数 aα= 10−7G 和 bα= 10−1G 在噪声精度分布中,其中 G 是基因总数。最后,在以下关联分析中包括 10 个 PEER 隐藏因子,以提高功效和可解释性。
表达数量性状位点的关联分析与鉴定
SNP 与基因表达水平之间的关联分析是使用 TASSEL v5 中实现的 MLM 进行的 (Bradbury et al., 2007)。前 3 个主成分和 10 个隐藏因子作为协变量,亲缘关系矩阵作为随机效应的方差-协方差矩阵。应用 BH 方法控制 α = 0.05 水平的 FDR。鉴定 eQTL 的方法与之前的研究相似 (Fu et al., 2013)。将距离小于 5 kb 的相邻 SNP 分组为一个簇,具有至少 3 个显著 SNP 的簇被视为候选 eQTL。如果单个基因的两个候选 eQTL 低于 LD (r2> 0.1),则显著性较低的 eQTL 被丢弃。如果两个候选 eQTL 具有相同的 eQTL 关联显著性 (P 值),则去除关节效应较小的 eQTL。
多靶点表达数量性状位点的鉴定和展示
对于每个 eQTL 区域,我们计算了其在两个发育阶段的靶标数量。如果 eQTLs 在 5 或 15 DAP 时与三个以上的基因相关,则将其定义为多靶点 eQTL。使用 Circos 软件可视化 eQTL 与其靶标之间的联系 (Krzywinski et al., 2009)。
核长度的关联分析
还使用 TASSEL v5 进行了籽粒长度的关联分析(Bradbury et al., 2007)。对于 MLM,前三个主成分用作协变量,亲缘关系矩阵用作随机效应的方差-协方差矩阵。对于 GLM,未包括协变量或随机效应。
用于检测核长度数量性状转录本的线性回归模型
我们使用以下线性回归模型来识别与内核长度相关的基因:
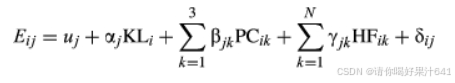
其中,Eij是自交系 I 中基因 j 的表达水平,uj是回归截距 KL我表示自交系 i 的内核长度,PC益(1≤ k ≤ 3) 表示自交系 i 的基因型的第 k 个主成分值,HF益(1≤ k ≤ N) 表示自交系 i 的基因表达谱第 k 个隐藏因子的值,N 是考虑最多的隐藏因子,δij是错误项,并且、αj, β开怀大吉, γ开怀大吉分别是核长度、第 k 个主成分和第 k 个隐藏因子的回归系数。在该模型中,我们考虑了前两个隐藏因素,它们对种群中的基因表达变异有广泛的影响。对于每个基因,如果 αj显著偏离 0,则认为基因 j 与籽粒长度有关。我们使用 P < 0.05 作为临界值,并定义基因 j 随表型上调,如果 αj> 0 并下调 αj< 0.
基因本体富集分析
使用在线工具包 agriGO 进行基因的 GO 富集分析(Du et al., 2010)。映射条目的最小数量设置为 5,并使用 Fisher 的精确检验来检查在相应全基因组 (AGPv3,Ensembl 版本 30) 背景下积累的意义。然后使用 Benjamini-Yekutieli 方法 (FDR < 0.05) 调整 P 值。
基序富集分析
为了搜索 mRNA 完整序列或 3’-UTR 区域上的基序分布,使用 Perl 编程语言中的正则表达式来匹配 cDNA 序列上已发表的基序。RRACH 基序的正则表达式是 [GA][GA]AC[TAC],而 URUAY 的正则表达式是 T[GA]TA[TA]。如果一个基因有多个转录本,当该基因的任何转录本在其 mRNA/3’-UTR 中包含基序时,我们会在其 mRNA/3’-UTR 中定义一个基序。在计算了包含基序的基因数量后,使用超几何函数计算基序富集的统计意义。
入藏号
5-DAP 籽粒中 282 个玉米自交系的 RNA 测序数据已提交给 NCBI 序列读取档案 (SRA;http://www.ncbi.nlm.nih.gov/sra/)替换为 BioProject ID PRJNA413629 或研究登录 ID SRP119998。处理后的基因表达丰度数据,包括读取计数和 FPKM 值,可以通过 NCBI 基因表达综合 (GEO;https://www.ncbi.nlm.nih.gov/geo/)与系列条目GSE110315。SNP 数据集已提交给欧洲变异档案馆 (EVA;https://www.ebi.ac.uk/eva),入藏号为 PRJEB24974。用于读取定位、基因表达分析和 SNP 检出的所有程序和脚本都可以在以下网站中找到:https://github.com/Pang-Junling/maize-5DAP-eQTL。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









