







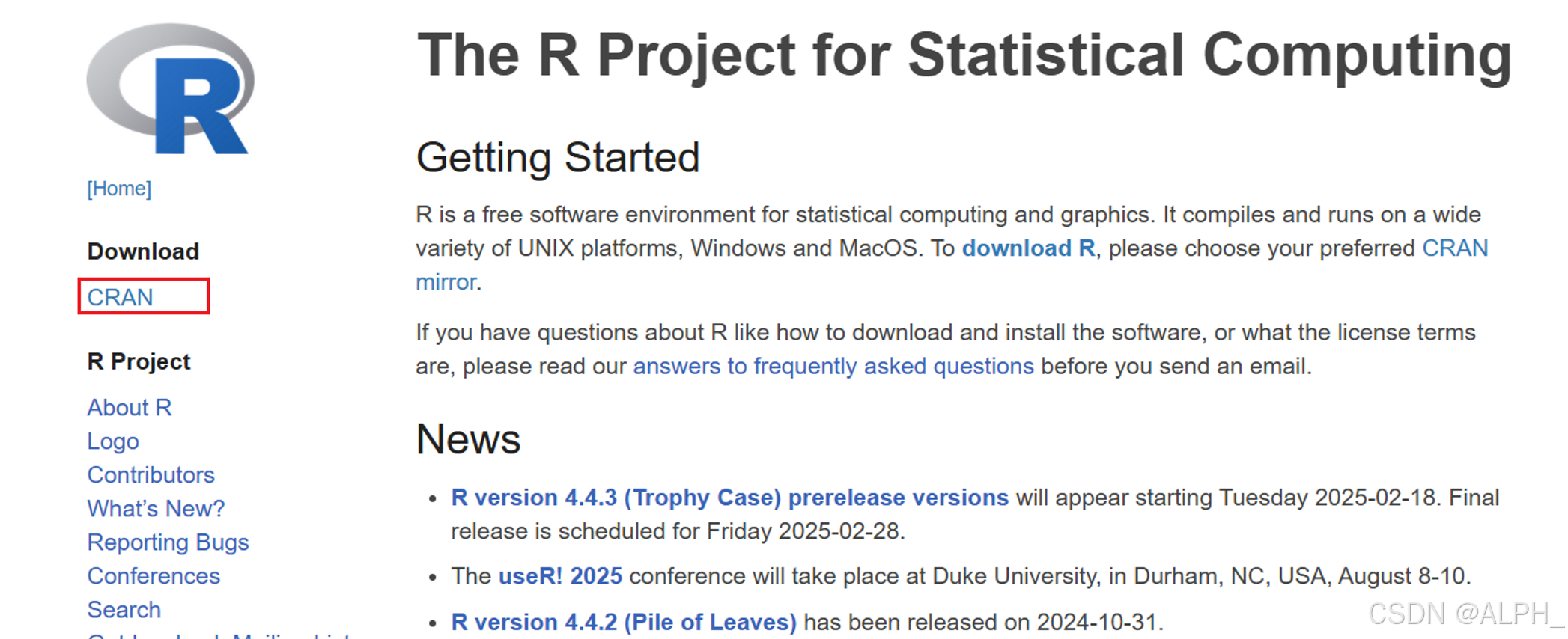
软件的安装:
r和rstudio
简单的加减:
> 1 + 1
[1] 2
> 2 ^ 10
[1] 1024
取范围-10到10:
> -10 : 10
[1] -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 0 1 2 3 4 5 6
[18] 7 8 9 10
赋值:
> x <- -10 : 10
> x
[1] -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 0 1 2 3 4 5 6
[18] 7 8 9 10
> x ^ 2
[1] 100 81 64 49 36 25 16 9 4 1 0 1 4 9 16 25 36
[18] 49 64 81 100
> y <- x ^ 2
> y
[1] 100 81 64 49 36 25 16 9 4 1 0 1 4 9 16 25 36
[18] 49 64 81 100
中位数:
> quantile(1 : 10)
0% 25% 50% 75% 100%
逻辑操作:
1.00 3.25 5.50 7.75 10.00
> 2 > 3
[1] FALSE
> 2 = 3
错误于2 = 3: (do_set)赋值公式左手无效
> 2 == 3
[1] FALSE
> 2 < 3
[1] TRUE
画散点图:
> x <- -10:10
> y <- x ^ 2
> plot(x,y)
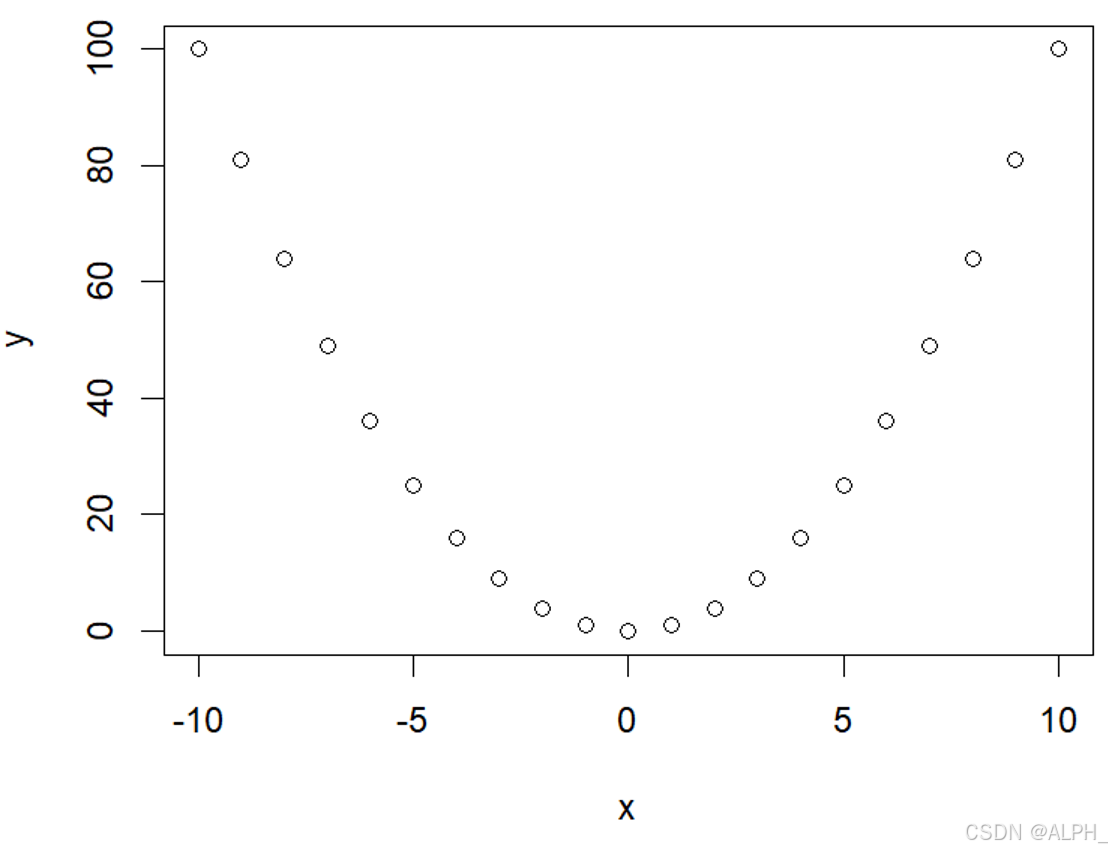
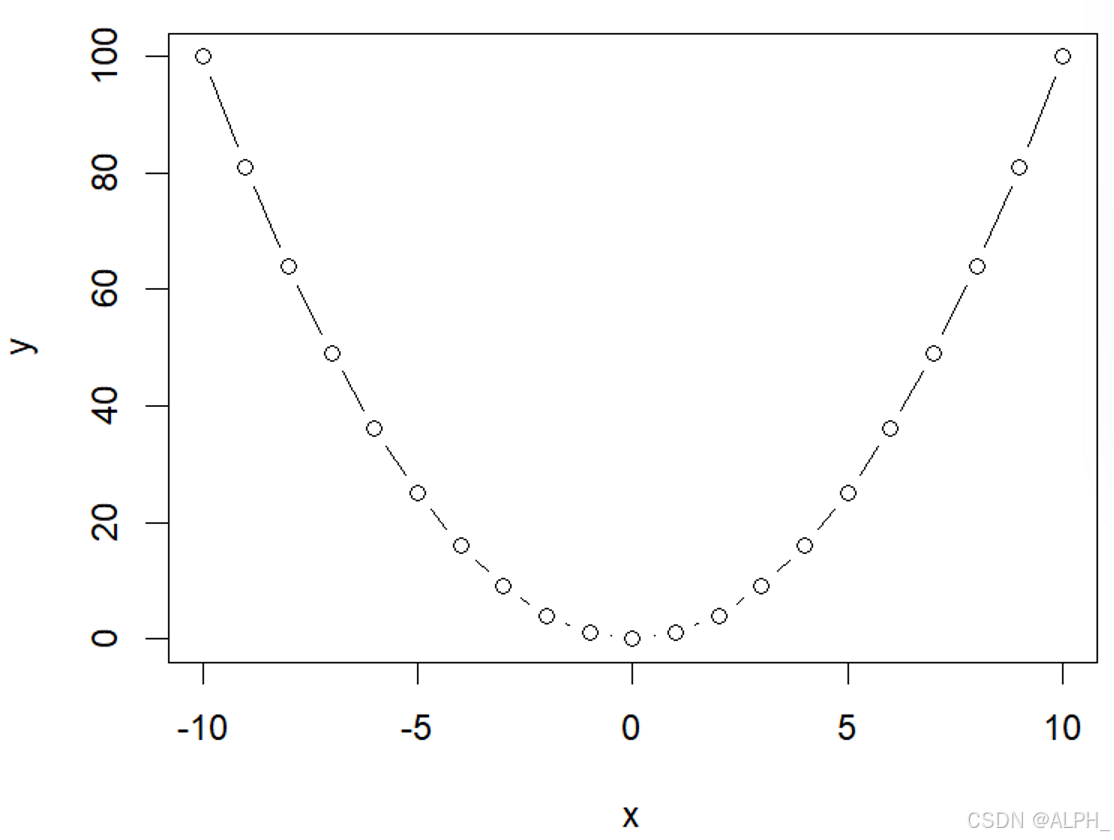
> plot(x,y,type = "b")
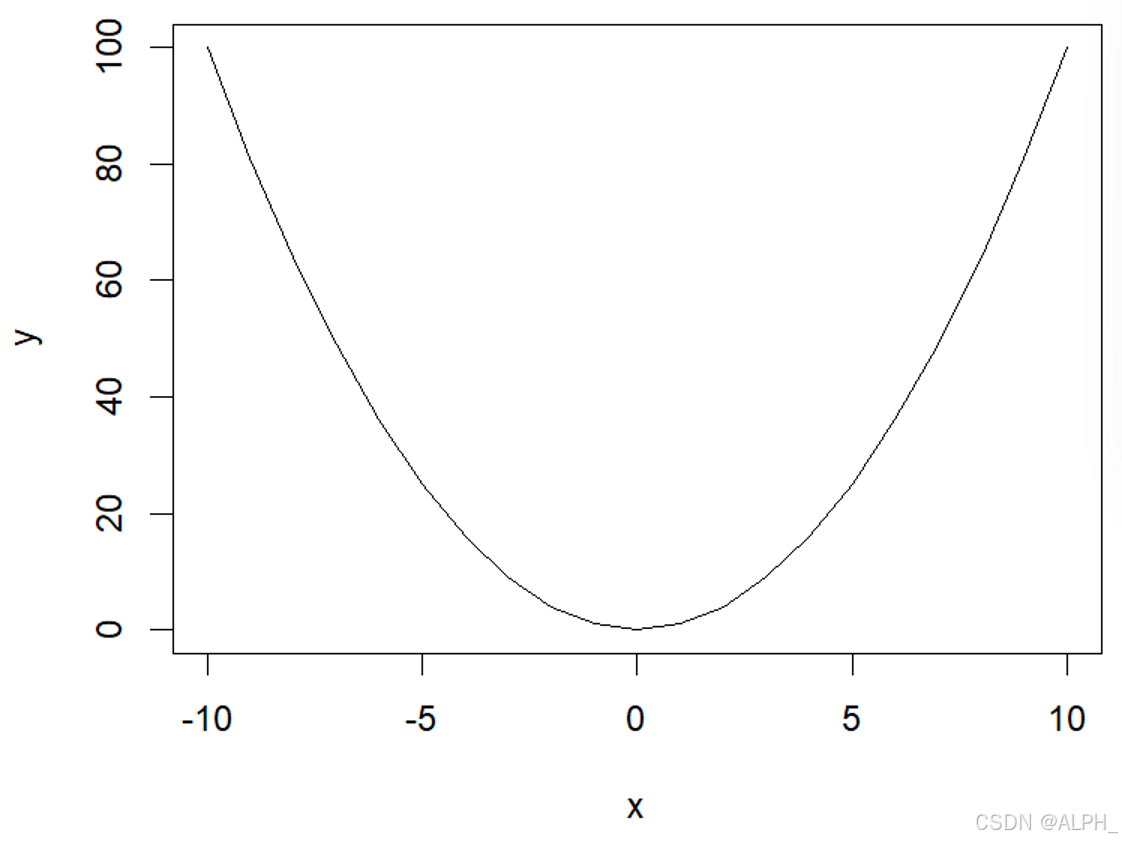
> plot(x,y,type = "l")
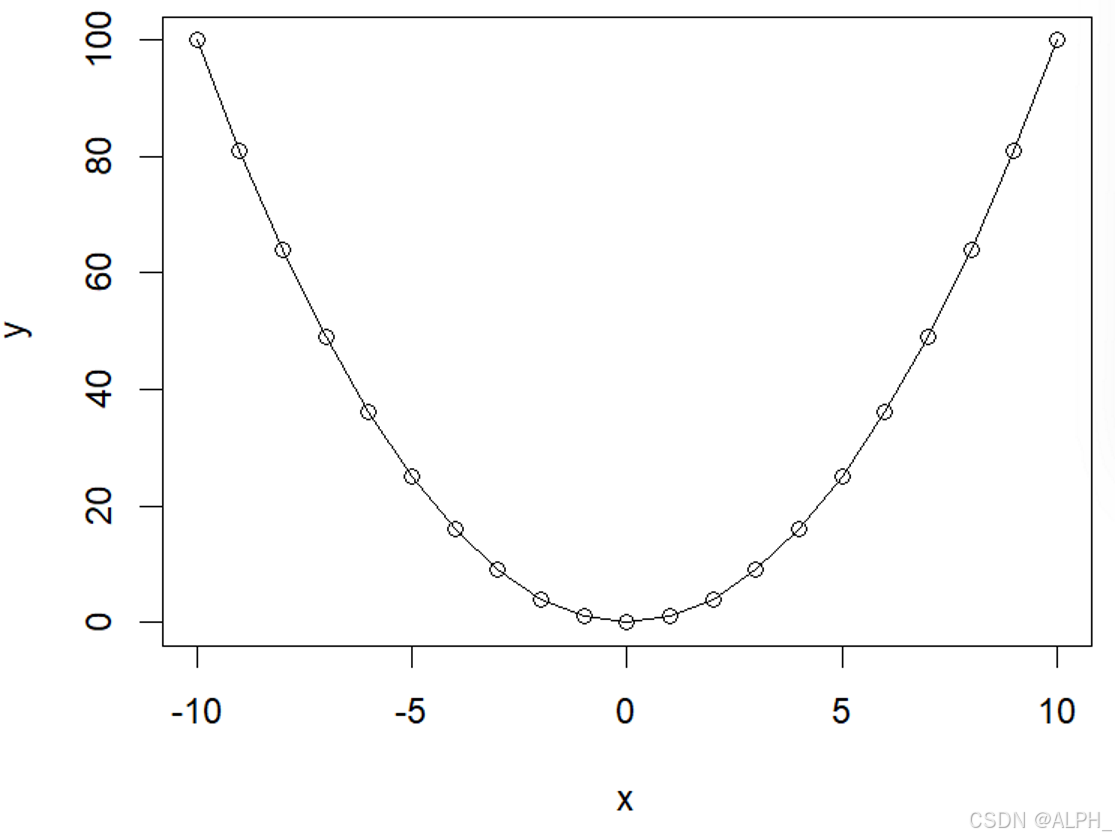
> plot(x,y,type = "o")
分别是下面三张图:
R包在生物信息学、统计分析以及系统发育与进化研究中有很多有用的功能:
- 基础功能包(Base):
- base:包含R的基本功能,所有R用户都会用到。
- 统计学包(Stats):
- stats:R的基础统计包,包含各种统计功能,如回归分析、t检验、方差分析等。
- 线性及非线性混合效应模型(Linear and Non-linear Mixed Effects Models):
- nlme:提供用于拟合线性和非线性混合效应模型的函数。
- 绘图(Graphics):
- graphics:R的基础绘图包,用于基本的2D图形绘制。
- ggplot2:用于创建复杂和定制化的图形,基于“语法图形学”的理念。
- lattice:用于创建多面板图形,特别适合探索高维数据的模式。
- 栅格图(Raster Graphics):
- raster:处理栅格数据的包,常用于空间数据的分析和绘图。
- 系统发育与进化分析:
- ape:用于系统发育分析的包,支持进化树的构建、操作和可视化。
- phytools:用于分析系统发育数据,包括树形比较和进化模型分析。
- apTreeshape:用于分析和比较系统发育树的形状。
- DNA序列分析(DNA Sequence Analysis):
- seqinr:提供DNA序列的读取、分析和操作功能。
- ade4:用于多元统计分析,适用于生态学和进化生物学数据分析。
- 生态学数据分析(Ecological Data Analysis):
- vegan:用于生态学数据分析,尤其是群落生态学和物种多样性研究。
- ecodist:提供生态学数据距离矩阵和多维尺度分析等功能。
- 其他生态学相关包:
- ecospat:进行物种分布模型分析。
- biomod2:进行生态模型建模的包,常用于物种分布预测。
- 聚类分析(Cluster Analysis):
- cluster:R中用于进行聚类分析的基本包,支持各种聚类方法如K-means、层次聚类等。
- factoextra:用于聚类分析的结果可视化,帮助解释聚类的结构。
- 广义加性模型(Generalized Additive Models):
- mgcv:用于拟合广义加性模型(GAM)和广义加性混合模型(GAMMs),广泛应用于非线性回归建模。
- 多变量分解(Multivariate Decomposition):
- mvpart:用于多变量分解,常用于分类和回归分析中的决策树模型。
- 线性及非线性混合效应模型(Linear and Non-linear Mixed Effects Models):
- nlme:用于拟合线性和非线性混合效应模型,支持复杂的随机效应和固定效应建模。
- 系统发育比较(Phylogenetic Comparison):
- ouch:用于进行基于进化树的系统发育比较分析,尤其适用于比较性状进化等问题。
- 差异表达基因筛选(Differential Gene Expression):
- limma:用于差异表达基因分析,适用于微阵列数据或RNA-Seq数据分析。
- DESeq2:RNA-Seq数据分析中常用的包,提供了对RNA-Seq数据的差异表达分析功能。
- RNA-Seq数据分析(RNA-Seq Analysis):
- edgeR:RNA-Seq数据分析,特别是差异表达分析,类似于DESeq2。
- DESeq2:同上,是RNA-Seq数据分析的主流包之一。
- 字符串操作(String Manipulation):
- stringr:一个强大的字符串操作包,提供了很多简洁的函数,用于文本数据处理和分析。
- 高级迭代循环(Advanced Iteration Loops):
- purrr:函数式编程包,提供了更多灵活的迭代功能,替代了基础R中的apply、lapply等函数。
- data.table:高效的循环和数据处理包,尤其在大数据集下效率较高。
- 数据操作(Data Manipulation):
- plyr:用于数据操作和处理,虽然dplyr包功能更强大且更常用,但plyr仍然是很多旧版代码的依赖。
包的本地下载:
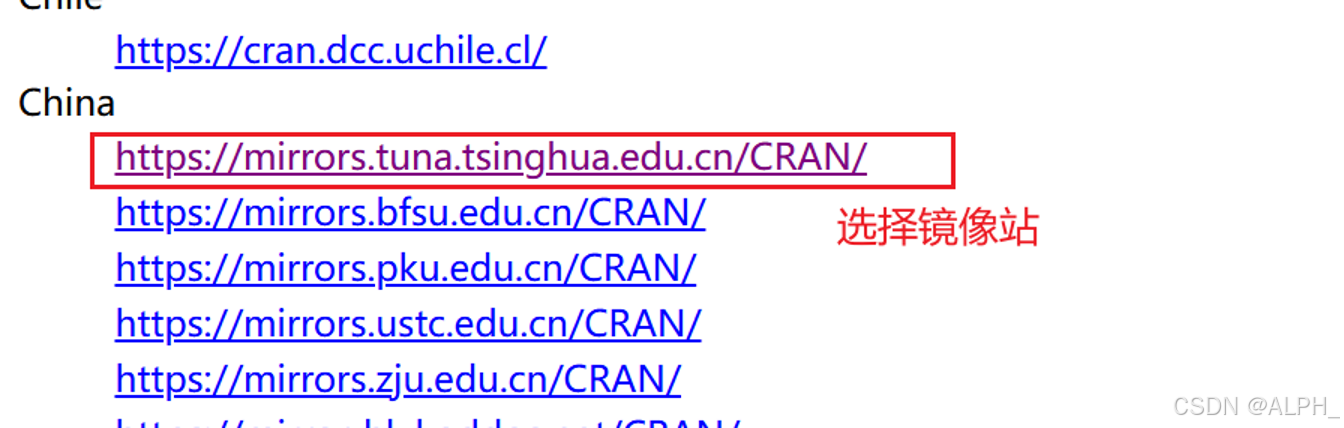
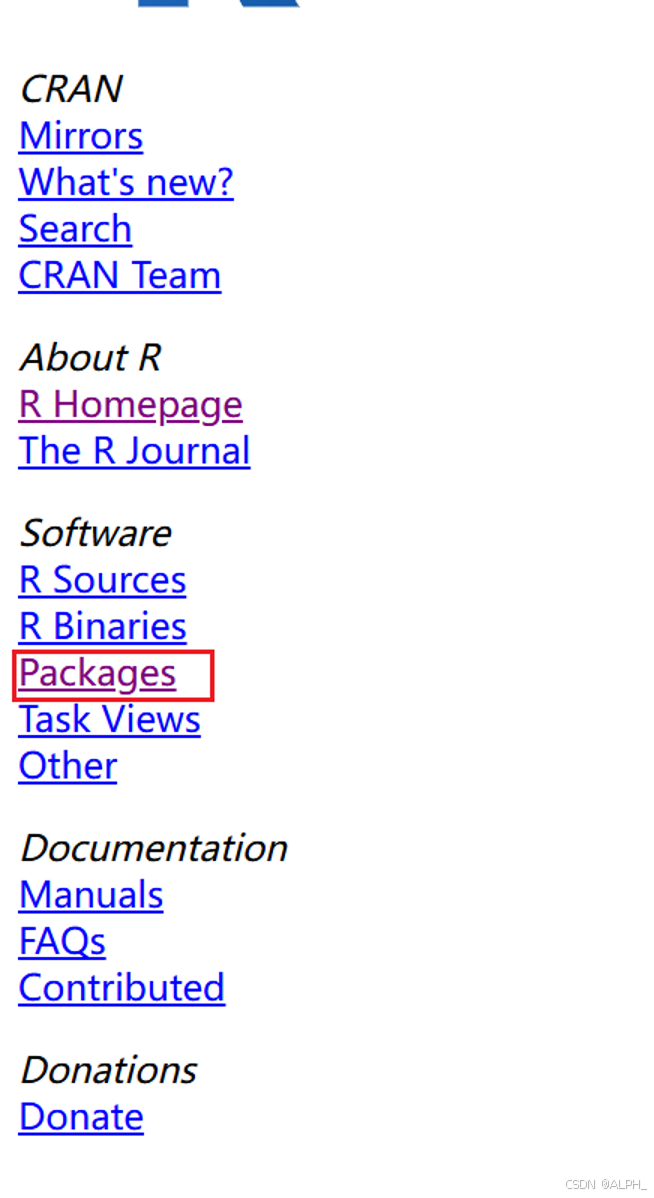

最下面这个task view就是根据这些所有的r包的能做的一些事情,它的一个特点。进行一个排序。
例如:

贝叶斯下面就是通过贝叶斯模型来进行统计分析的这些包

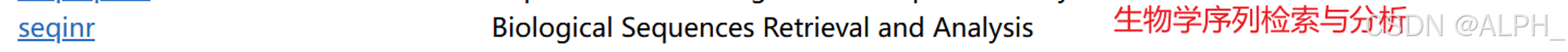

安装后不要解压缩,直接用r打开
安装方法(两种):
1:
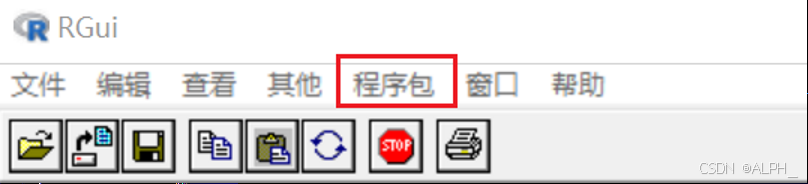
选择镜像,后选择包,就会自动安装,依赖包也会自己安装
2:
install.packages("包名")
关于Bioconductor:
Bioconductor 是一个独立于 CRAN(R 的主要包仓库)的 R 包仓库,专门用于存储与生物信息学相关的 R 包。它主要面向高通量测序(high throughput sequencing)、基因组学数据分析等领域,提供了大量处理生物数据的工具。像 limma(用于差异表达分析)、DESeq2(RNA-Seq 数据分析)、edgeR(RNA-Seq 数据分析)、以及用于处理单核苷酸多态性(SNP)、拷贝数变异(CNV)分析等方面的包,都可以在 Bioconductor 上找到。
Bioconductor 是一个开源项目,所有的包都是用 R 语言编写的,旨在为生物信息学领域的研究人员提供强大且灵活的数据分析工具。这些包涵盖了从数据预处理、分析、可视化到结果解读等各个方面,支持的功能包括基因表达分析、基因组数据分析、基因组序列比对等。
如果你对高通量数据分析有需求,Bioconductor 提供的包几乎是必不可少的资源,尤其是在处理大规模基因组数据时。

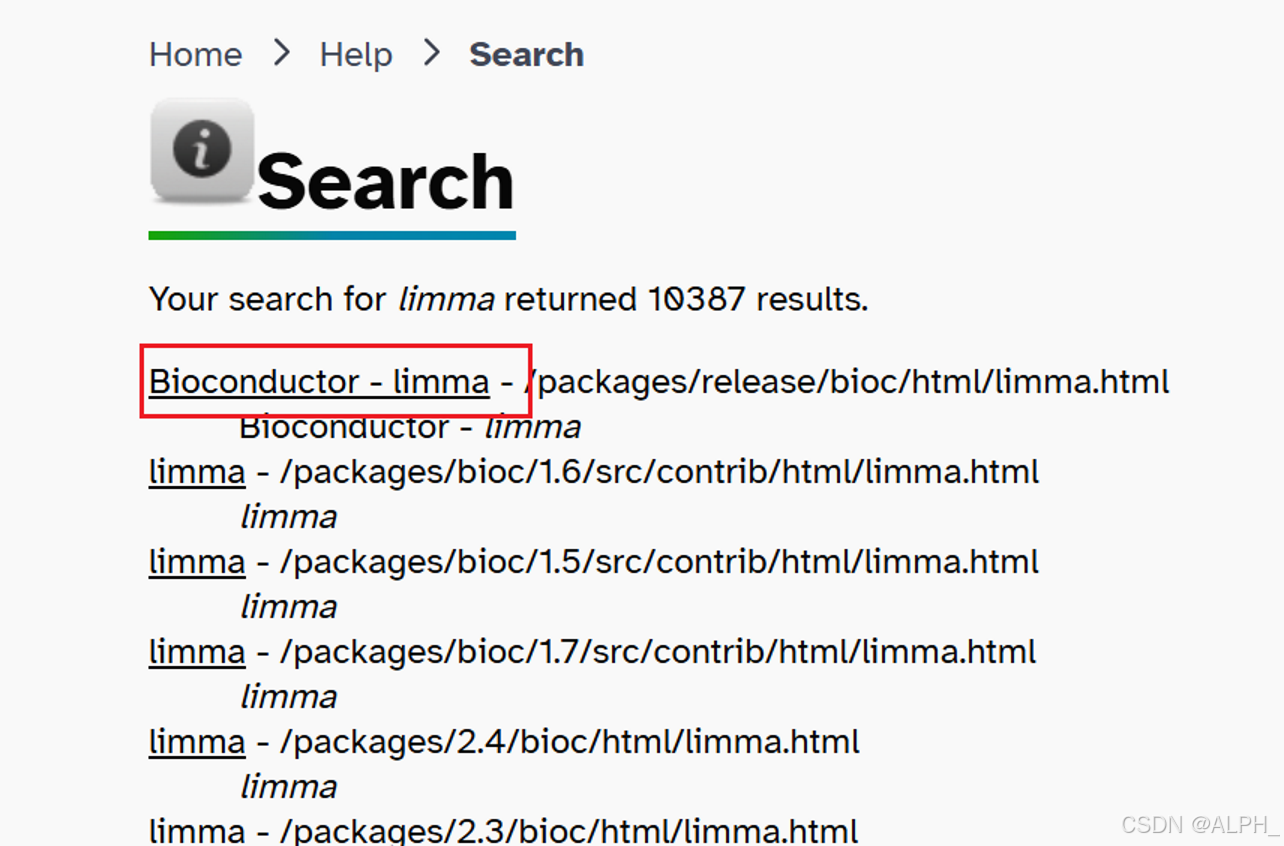


下载:

命令行安装:
if (!require("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("limma")


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









