









第一次开发类似于生物化学的程序,基于网络服务,使用JAVA语言编写,昨天总算初步完成,我在国内看类似的东西好像不多,我贴几个我程序图片,欢迎同行点评。
主窗口
加载要查询的基因列表
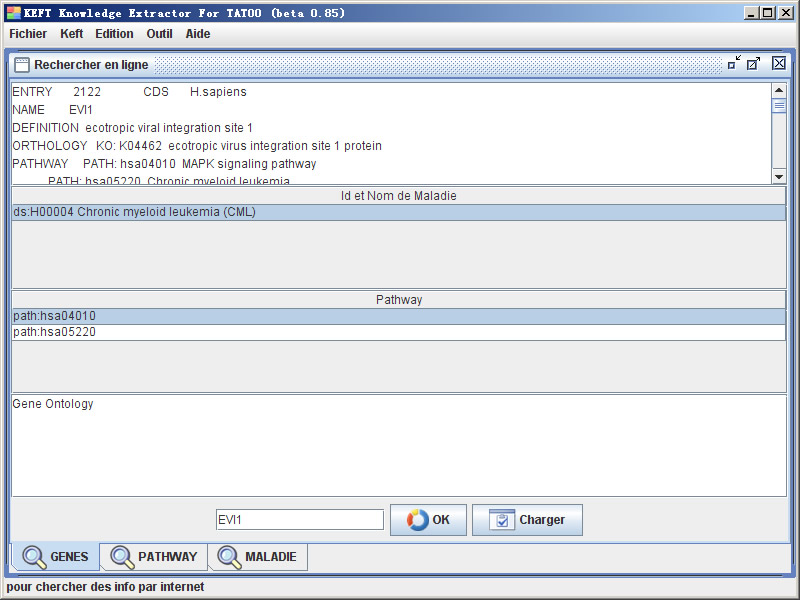
基因查询结果
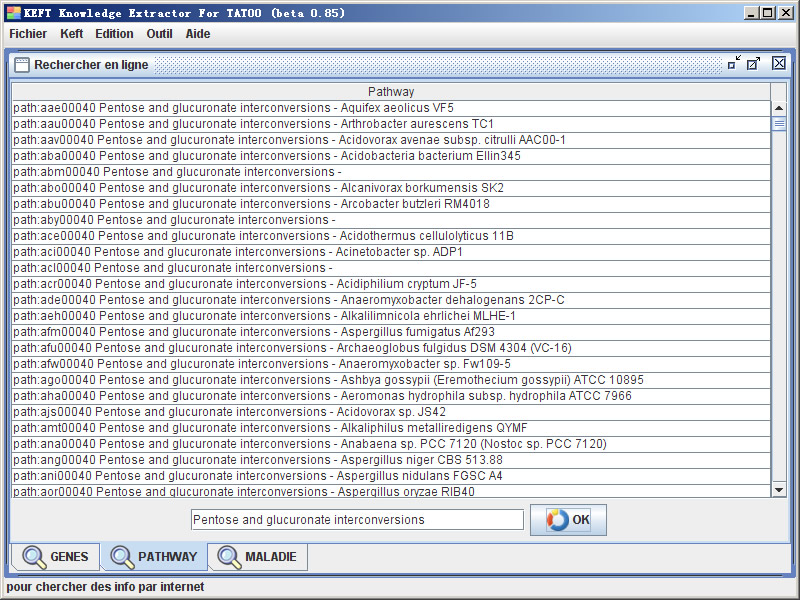
路径搜索结果
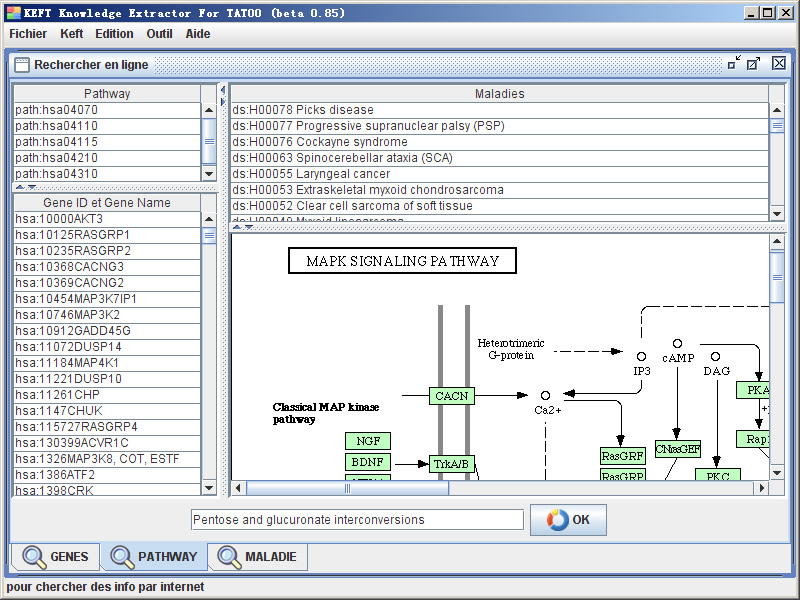
路径搜索结果
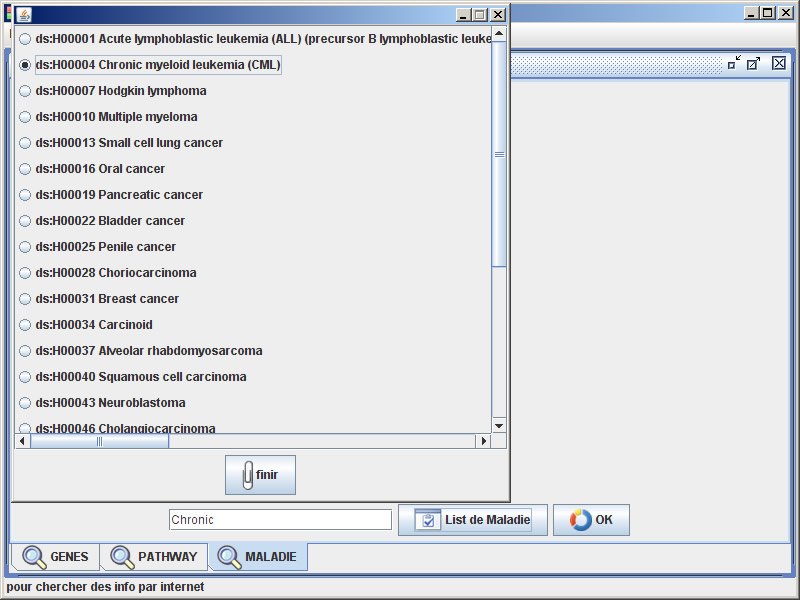
疾病搜索引擎
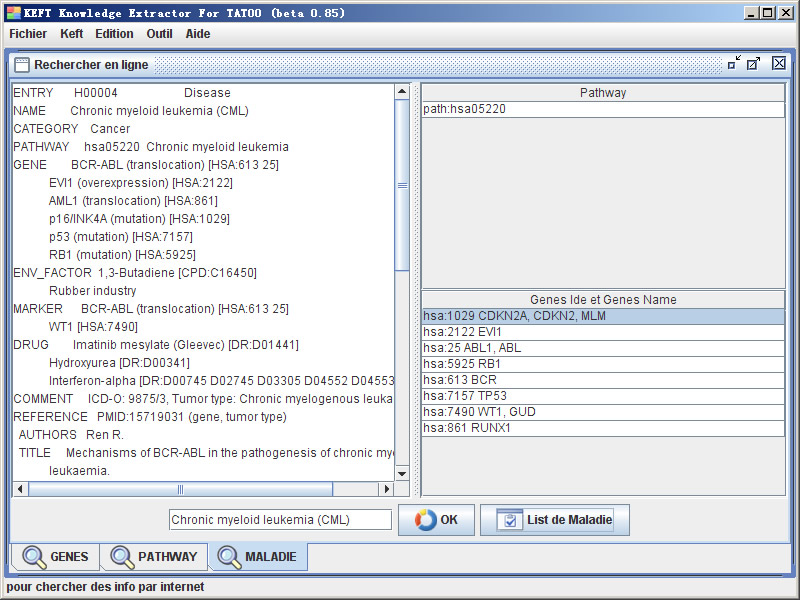
疾病搜索结果展示
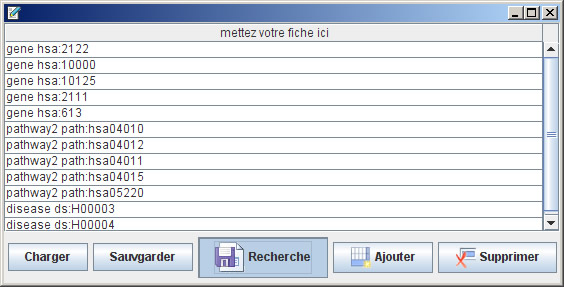
编辑器
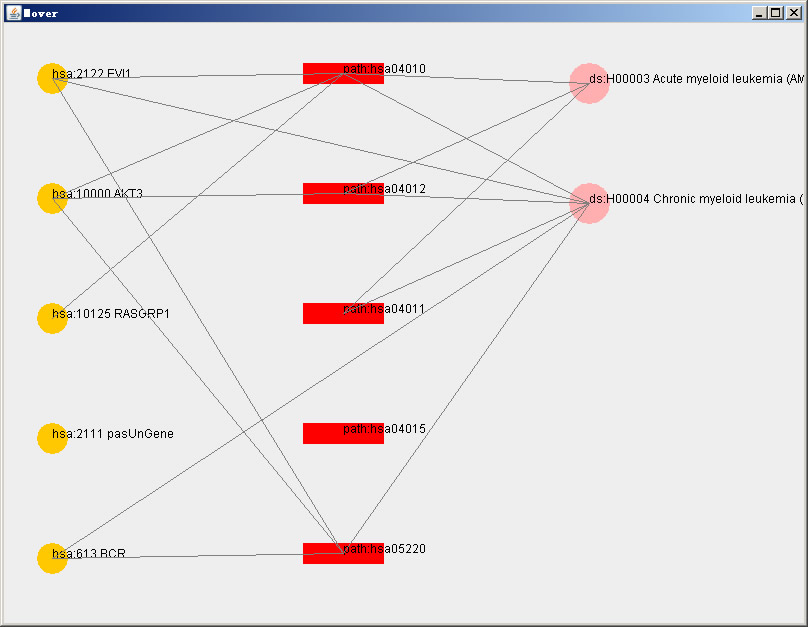
图像表示深层关系查找,每一个元素都可以移动。

第一次开发类似于生物化学的程序,基于网络服务,使用JAVA语言编写,昨天总算初步完成,我在国内看类似的东西好像不多,我贴几个我程序图片,欢迎同行点评。
主窗口
加载要查询的基因列表
基因查询结果
路径搜索结果
路径搜索结果
疾病搜索引擎
疾病搜索结果展示
编辑器
图像表示深层关系查找,每一个元素都可以移动。
 639
601
1031
722
639
601
1031
722

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言
























