







Ma, A., Wang, X., Li, J. et al. Single-cell biological network inference using a heterogeneous graph transformer. Nat Commun 14, 964 (2023).
论文地址:https://doi.org/10.1038/s41467-023-36559-0
代码地址:https://github.com/OSU-BMBL/deepmaps
摘要
单细胞多组学(scMulti-omics)能够同时量化多种模式,以捕捉复杂分子机制和细胞异质性的精妙之处。然而,现有工具无法有效推断不同细胞类型中的活性生物网络及其对外界刺激的响应。为此,我们提出了DeepMAPS,一种用于从scMulti-omics中推断生物网络的工具。DeepMAPS将scMulti-omics建模为异构图,利用多头图变换器(graph transformer),在局部和全局上下文中以鲁棒的方式学习细胞与基因之间的关系。
基准测试结果表明,DeepMAPS在细胞聚类和生物网络构建方面优于现有工具。在肺部肿瘤白细胞的CITE-seq数据以及匹配的弥漫性小淋巴细胞淋巴瘤scRNA-seq和scATAC-seq数据中,DeepMAPS还展现了其推断特定细胞类型生物网络的卓越能力。此外,我们还部署了一个DeepMAPS网页服务器,提供多种功能和可视化工具,以提升scMulti-omics数据分析的可用性和可重复性。
引言
单细胞测序技术(如单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq))正在重新塑造对细胞异质性的研究,并为神经科学、癌症生物学、免疫肿瘤学和治疗反应等领域带来重要见解。然而,单一的单细胞模式仅能反映遗传特征的一个片段,无法全面描绘细胞的特性,从而导致复杂生物系统表征的偏差。单细胞多组学(scMulti-omics)技术能够同时量化多种模式,从而全面捕捉复杂分子机制和细胞异质性的细微差别。当这种技术与稳健的计算分析方法结合时,可大幅推进各种生物学研究。
现有用于整合分析scMulti-omics数据的工具(如Seurat、MOFA+、Harmony和totalVI)能够可靠地预测细胞类型和状态、去除批次效应,并揭示不同模式之间的关系或对齐。然而,大多数方法未明确考虑细胞和模式之间的拓扑信息共享。因此,这些方法难以同时推断出多种细胞类型的活性生物网络,并在特定细胞类型中揭示复杂网络对外界刺激的响应。
近年来,图神经网络(GNN)在通过传播邻近细胞特征和构建全局细胞图来学习单细胞的低维表示方面表现出色。例如,我们团队开发的scGNN工具(基于大规模scRNA-seq数据的GNN模型)在细胞聚类和基因推补性能上表现优越。此外,异构图(由不同类型的节点和边构成的图)已被广泛用于建模多关系知识图谱,它为整合scMulti-omics数据并学习特定细胞类型的生物网络提供了自然的表示框架。同时,注意力机制的最新发展使得异构关系建模更加高效,不仅提升了深度学习模型的可解释性,还促进了特定细胞类型生物网络的推断。
在本研究中,我们开发了DeepMAPS(基于深度学习的单细胞多组学分析平台),一个用于从scMulti-omics数据中推断特定细胞类型生物网络的异构图变换器框架。该框架基于先进的GNN模型(异构图变换器,HGT),具有以下优势:
1. 构建了一体化的异构图,其中将细胞和基因作为节点,将它们之间的关系作为边。
2. 模型同时捕捉了细胞和基因之间的邻近和全局拓扑特征,用以构建细胞-细胞和基因-基因关系。
3. HGT模型中的注意力机制可以评估基因对特定细胞的重要性,从而区分基因贡献并增强生物学解释性。
4. 模型无需依赖基因共表达约束,能够推断其他工具通常无法识别的基因调控关系。
值得一提的是,DeepMAPS被实现为一个无需编码的交互式界面,并配备了Docker工具包,降低了用户在scMulti-omics数据分析中的编程负担,从而进一步提升了其可用性和便捷性。
模型概述
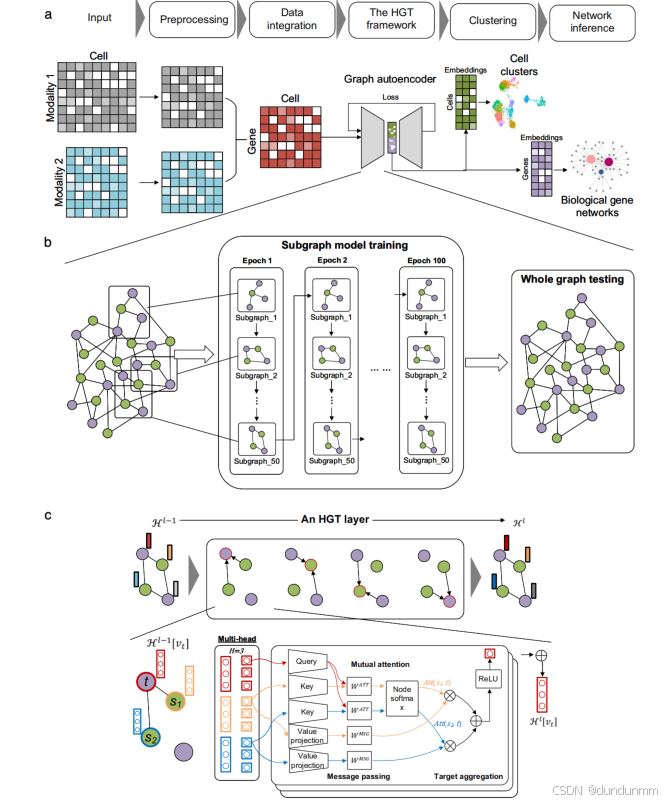
总体而言,DeepMAPS是一个端到端、无需假设的框架,用于从单细胞多组学数据中推断特定细胞类型的生物网络。DeepMAPS 框架包含五个主要步骤(见图 1 和方法部分):
1. 数据预处理:去除低质量细胞和低表达基因,并根据具体数据类型应用不同的归一化方法。生成一个综合的细胞-基因矩阵,用于表示每个基因在每个细胞中的综合活性。针对不同类型的单细胞多组学数据,应用特定的数据整合方法。
2. 构建异构图:基于综合矩阵构建异构图,图中包括细胞和基因作为节点,基因在细胞中的存在作为边。
3. 构建 HGT 模型:利用 HGT(异构图变换器)模型,联合学习细胞和基因的低维嵌入,并生成注意力分数,指示基因对细胞的重要性。
4. 细胞聚类与功能基因模块预测:基于 HGT 学习的嵌入和注意力分数进行细胞聚类及功能基因模块预测。
5. 推断多样的生物网络:例如,针对每种细胞类型推断基因调控网络(GRN)和基因关联网络。
框架工作流程
为了学习细胞和基因的联合表示,首先从输入的单细胞多组学数据生成细胞-基因矩阵,整合相关信息。接着构建异构图,其中细胞节点和基因节点通过未加权的细胞-基因边连接,表示基因在细胞中的活性存在情况。每个节点的初始嵌入通过两层图神经网络(GNN)图自动编码器从细胞-基因综合矩阵中学习得出。此异构图可以清晰地表示并有机整合单细胞多组学数据,从而协同学习出具有生物学意义的特征。
整个异构图被输入图自动编码器,用于学习细胞和基因之间的关系,并更新每个节点的嵌入。DeepMAPS 采用一种异构多头注意力机制,在异构图上建模全局拓扑信息(全局关系)和邻域信息传递(局部关系)。异构图表示学习为同时嵌入细胞和基因提供了一种方式,并通过 DeepMAPS 的变换器实现。
注意力机制
在每一层 HGT 中,将每个节点(细胞或基因)视为目标节点,其一跳邻居视为源节点。DeepMAPS 通过节点嵌入的协同作用(即注意力分数)评估邻居节点的重要性以及传递给目标节点的信息量。最终,嵌入高度正相关的细胞和基因更有可能彼此传递信息,从而最大化嵌入的相似性和差异性。
分布式子图训练
为了在大规模异构图上实现可行的无监督训练,DeepMAPS 对异构图随机采样 50 个子图进行操作,每个子图覆盖至少 30% 的细胞和基因,用于共享参数的训练。这些参数随后用于测试整个图。
训练结果
作为训练的重要成果,注意力分数反映了基因对细胞的重要性。基因对细胞的高注意力分数表明该基因在定义细胞身份和表征细胞异质性方面相对重要。这种区分性允许在每个细胞簇中构建可靠的基因关联网络,作为 DeepMAPS 的最终输出。
基因关联网络构建
随后使用斯坦纳森林问题(SFP)模型来识别对某一细胞簇具有较高注意力分数和相似嵌入特征的基因。SFP 模型优化解中的基因-基因和基因-细胞关系反映了基因嵌入的相似性和基因对细胞簇的重要性。通过基于注意力分数和嵌入相似性筛选出对细胞簇特征表征最重要的基因,从而建立基因关联网络。这些基因被认为是细胞类型活跃基因(cell-type-active genes)。
讨论
DeepMAPS 是一种深度学习框架,通过实现异构图表示学习和图变换器,用于从单细胞多组学数据中研究生物网络。通过构建包含细胞和基因的异构图,DeepMAPS 同时识别其联合嵌入,并在一个完整的框架中推断特定细胞类型的生物网络及其对应的细胞类型。此外,应用异构图变换器以可解释的统一多关系形式建模细胞与基因之间的关系,从而在训练和学习过程中显著缩短了图中远距离细胞影响的计算时间。
通过联合分析基因表达和蛋白丰度,DeepMAPS 准确识别并注释了混合 PBMC 和肺肿瘤白细胞 CITE-seq 数据中的 13 种细胞类型,基于精心挑选的标记,这些特征单一模式无法完全解释。此外,DeepMAPS 所识别的嵌入特征能够在原始信号嘈杂的情况下捕获并放大统计上显著的信号。我们还基于两组簇推断的基因关联网络,识别了 DC 和 TRM CD4+ T 细胞之间具有生物学意义的细胞-细胞通信途径。对于 scRNA-ATAC-seq 数据,我们采用了一种基于 RNA velocity 的方法,动态整合基因表达和染色质可及性,增强了细胞簇预测能力。利用此方法,我们识别了正常 B 细胞与两种 DSLL 发育状态之间的不同基因调控模式,并进一步揭示了细胞-细胞通信与下游基因调控网络(GRNs)之间的深层生物学联系,这有助于表征和定义 DSLL 状态。此外,所识别的转录因子(TFs)和基因可能成为进一步验证和治疗 DSLL 的免疫治疗靶点。
尽管 DeepMAPS 在分析单细胞多组学数据方面具有优势并表现出改进的性能,但仍有进一步优化的空间。首先,对于超大规模数据集(如超过 100 万个细胞),异构图表示的复杂性(可能包含数十亿条边)可能带来计算效率的问题。此外,建议在 GPU 上运行 DeepMAPS,但这可能导致可重复性问题。不同的 GPU 模型因浮点数精度差异可能影响训练过程中的损失函数精度,进而导致不同 GPU 模型生成的细胞聚类和网络结果略有差异。最后,当前版本的 DeepMAPS 基于包含基因和细胞的二部异构图,要求在整合到细胞-基因矩阵之前对不同模式进行单独的预处理和整合步骤。为了实现完全的端到端单细胞多组学分析框架,可以将二部图扩展为多部异构图,将不同的模式作为不相交的节点类型(如基因、蛋白质或峰区域)纳入其中。此外,这种多部异构图还可以包括基于知识的生物学信息,例如已知的分子调控,以及在单个图中整合两个以上的模式。然而,随着节点类型的增加,计算负担将呈几何增长,这需要未来在模型和参数优化方面进行专门研究。
综上所述,我们评估了 DeepMAPS 作为整合分析单细胞多组学数据和推断特定细胞类型生物网络的开创性研究。该框架为深度学习在单细胞生物学中的应用提供了不同的视角。随着 DeepMAPS 网络服务器的开发与维护,我们的长期目标是创建一个基于深度学习的生态社区,用于存档、分析、可视化和传播 AI 就绪的单细胞多组学数据。
后续将详细讨论模型的具体实现和实验结果分析。异构图是想法最近已经看到几篇了,确实是一个值得思考的方向。。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









