






前言
DNA甲基化的富集分析不同于GWAS,因为CpG位点在每个基因或基因启动子之间的分布是不均衡的,不同基因对应的探针数也有很大的差异,所以不能直接用“cluster Profiler”来做GO分析和KEGG分析。
那DNA甲基化的富集分析用什么工具呢?Bingo~答案就是文章标题中的“missMethyl”R包了。它可以通过加权重抽样和Wallenius非中心超几何近似法矫正“多基因偏差”和“多探针偏差”。下面是相关的代码。
missMethyl R包进行甲基化数据的富集分析
1.missMethyl 的安装
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("missMethyl")
library(missMethyl)R的版本要在4.3以上。这一步会比较慢,需要耐心等待。
2.数据准备
missMethyl不需要输入基因的Entrez ID,直接输入差异甲基化的探针名称(sig.cpg)和所有的探针名称(all.cpg)即可。
#差异甲基化探针的名称存到sig.cpg当中
sig.cpg<-as.vector(sig$DNAm)
#所有探针的名称存到all.cpg当中
all.cpg<-rownames(methy_map)3.调用gometh()函数进行富集分析
#GO分析
gst <- gometh(sig.cpg = sig.cpg,all.cpg=all.cpg,collection = "GO",
plot.bias = TRUE,prior.prob = TRUE,array.type = "EPIC")
table(gst$FDR<0.05)
GO.sig<-topGSA(gst)
write.csv(GO.sig,'GO.sig.csv')
#KEGG分析
kegg <- gometh(sig.cpg = sigcpg,all.cpg=allcpg,collection = "KEGG",
plot.bias = TRUE,prior.prob = TRUE,array.type = "EPIC")
table(kegg$FDR<0.05)
KEGG.sig<-topGSA(kegg)
write.csv(KEGG.sig,'KEGG.sig.csv')collection='GO'表明进行GO分析,collection='KEGG'表明进行KEGG分析,plot.bias=TRUE则会显示每个基因中探针的数量和差异甲基化的概率之间的关系,prior.prob如果为真,将考虑由于每个基因的探针数量而导致显着差异甲基化的概率。如果为 false,则执行超几何检验,忽略数据中的任何偏差。array.type=c("450K","EPIC"),默认值为450K,代码中的是“EPIC”芯片。
接着,使用table( )函数查看FDR值<0.05的结果,最后将显著的结果保存成.csv文件。
4.结果展示
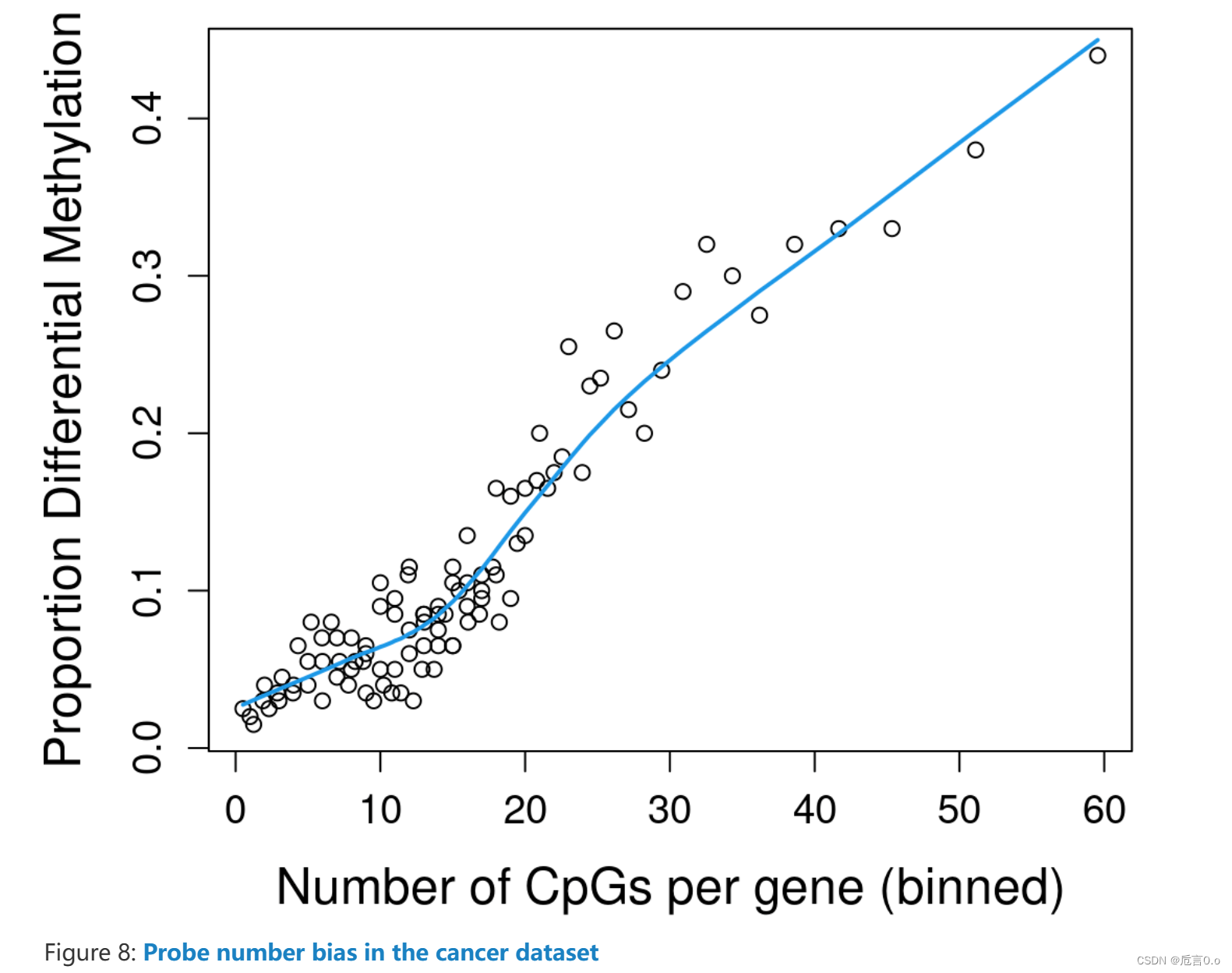
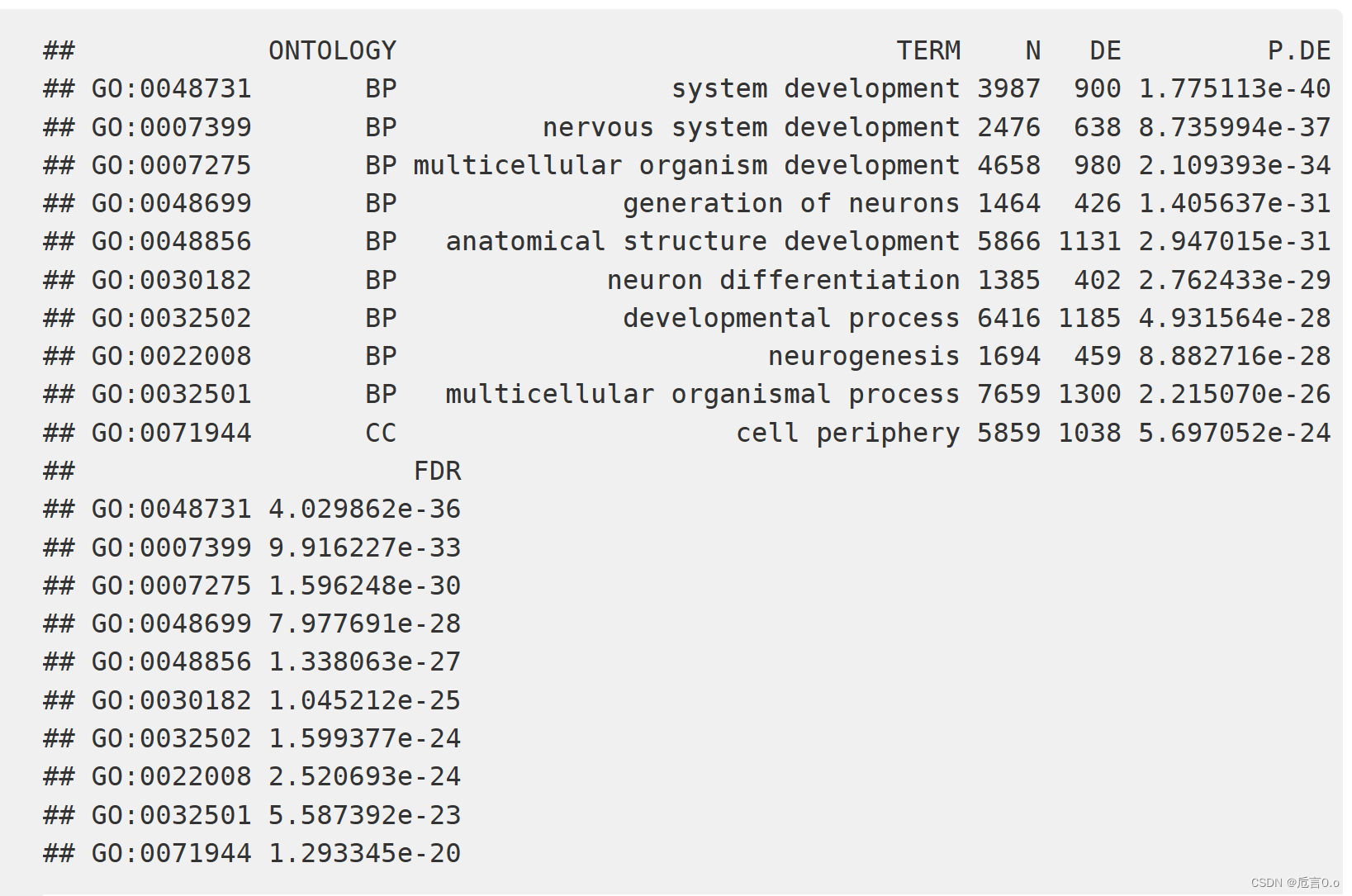
左图是每个基因中探针的数量和差异甲基化的概率之间的关系图,右图是GO分析的结果,N表示number of genes in the GO or KEGG term,DE表示number of genes that are differentially methylated。
以上就是这篇文章的全部内容了~由于作者本人也是个初学者,难免水平不足,如果文章内容有错误或表述不准确的地方,欢迎大家在评论区批评指正。另外,希望了解更多有关missMethyl R包的友友们,可以查看官方的文档(missMethyl: Analysing Illumina HumanMethylation BeadChip Data (bioconductor.org))。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









