






标题:Review of Single-cell RNA-seq Data Clustering for Cell Type Identification and Characterization
单细胞RNA-seq数据聚类用于细胞类型鉴定和表征的综述
作者: Shixiong Zhanga, Xiangtao Lia, Qiuzhen Linaand Ka-Chun Wonga
关键字: Single-cell RNA-seq(单细胞RNA测序) 、Clustering(聚类)、Cell types(细胞类型)、Identification(识别)、Characterization(特征)、Review(综述、回顾)
文献PDF下载:https://download.csdn.net/download/lesileqin/18979322
文章目录
Abstract 摘要
In recent years, the advances in single-cell RNA-seq techniques have enabled us to perform large-scale transcriptomic profiling at single-cell resolution in a high-throughput manner. Unsupervised learning such as data clustering has become the central component to identify and characterize novel cell types and gene expression patterns.
近年来,单细胞RNA-seq技术的发展使我们能够在单细胞分别率和高通量的方式下进行大规模转录组分析。无监督学习,比如数据聚类,已经成为识别和特征新的细胞类型和基因表达模式的中心组件。
In this study, we review the existing single-cell RNA-seq data clustering methods with critical insights into the related advantages and limitations. In addition, we also review the upstream singlecell RNA-seq data processing techniques such as quality control, normalization, and dimension reduction. We conduct performance comparison experiments to evaluate several popular single-cell RNA-seq clustering approaches on two single-cell transcriptomic datasets.
在本研究中,我们回顾了现有的单细胞RNA-seq数据聚类的方法,并对相关的优点和局限性进行了关键的见解。此外,我们还回顾了上游单细胞RNA-seq数据处理技术,如质量控制、归一化和降维。我们在两个单细胞转录组数据集上进行了性能比较实验,以评估几种流行的单细胞RNA-seq聚类方法。
1. Introduction 介绍
With the unabated progress in high-throughput sequencing technologies, single-cell RNA-seq has become a powerful approach to simultaneously measure cell-to-cell expression variability of thousands or even hundreds of thousands of genes at single cell resolution. Such high-throughput transcriptomic profiling can capture the gene transcriptional activities to reveal cell identities and functions and discover cell types or even rare cell types. Hence, one of the most common goals of those single-cell studies is to identify cell subpopulations under different contexts. The gene expression patterns of those subpopulations help us distinguish various cell types and functions, identifying different cell types.
随着高通量测序技术的不断进步,单细胞RNA-seq已经成为一种强大的方法,可以在单细胞分辨率下同时测量数千甚至数十万个基因在细胞间的表达变化。这种高通量转录组分析可以捕获基因转录活动,以揭示细胞身份和功能和发现细胞类型,甚至稀有细胞类型。因此,这些单细胞研究最常见的目标之一是识别不同环境下的细胞亚群。这些亚群的基因表达模式帮助我们区分不同的细胞类型和功能,识别不同的细胞类型。
Diverse computational approaches based on data clustering have emerged to interpret and understand single-cell RNA-seq data. The advances in single-cell clustering has also initiated the development of multiple atlas projects such as Mouse Cell Atlas, Aging Drosophila Brain Atlas, and Human Cell Atlas. However, several technical challenges are still involved in single-cell RNA-seq clustering. Low-quality cells/genes, amplification biases, and other confounding factors can affect the downstream clustering performance. In addition, given the whole transcriptome range of RNA-seq the curse of dimensionality should be expected. Thus the data preprocessing steps including quality control, normalization, and dimensional reduction have become necessary before downstream interpretation.In addition, the tissue heterogeneity can also affect the single-cell RNA-seq clustering performance to detect rare cell types.
基于数据聚类的多种计算方法已经出现来解释和理解单细胞RNA-seq数据。单细胞集群研究的进展也启动了多个图谱项目的开发,如小鼠细胞图谱、衰老果蝇大脑图谱和人类细胞图谱。然而,在单细胞RNA-seq聚类中仍然存在一些技术挑战。低质量的细胞或基因、扩增偏差和其他混杂因素会影响下游聚类性能。 此外,鉴于RNA-seq的整个转录组范围,维数灾难是可以预料的。在下游互连之前,包括质量控制、归一化和降维在内的数据预处理步骤已经成为必要。此外,组织异质性也会影响检测罕见细胞类型的单细胞RNA-seq聚类性能。
In this study, we review the recently developed computational clustering approaches for understanding and interpreting single-cell RNA-seq data. We also review the upstream single-cell RNA-seq data preprocessing steps such as quality control, row/column normalization, and dimension reduction before clustering is performed. Four roughly-classified categories of single-cell RNA-seq clustering methods and its application are discussed in terms of the strengths and limitations, including k-means clustering, hierarchical clustering, community-detection-based clustering, and densitybased clustering. Figure 1 depicts the workflow of single cell RNA-seq data clustering by data processing (quality control, normalization, and dimension reduction) and clustering methods. The strengths and limitations are discussed in following sections to guide selection of different tools. In addition, we conduct several experiments on single-cell RNA-seq datasets to evaluate and compare those clustering methods.
在这项研究中,我们回顾了最近开发的用于理解和解释单细胞RNA-序列数据的计算聚类方法。我们还回顾了上游单细胞RNA-seq数据预处理步骤,如质量控制、行/列标准化和聚类前降维。本文讨论了四种粗略分类的单细胞RNA-序列聚类方法及其应用,包括k-均值聚类、层次聚类、基于社区检测的聚类和基于密度的聚类。下图描述了通过数据处理(质量控制、标准化和降维)和聚类方法进行单细胞RNA-seq数据聚类的工作流程。以下各节将讨论其优势和局限性,以指导不同工具的选择。此外,我们在单细胞RNA-seq数据集上进行了几个实验来评估和比较这些聚类方法。
即本文对四种`聚类方法`进行了讨论,四种聚类方法分别是:
- k均值聚类
- 层次聚类
- 基于社区检测的聚类
- 基于密度的聚类
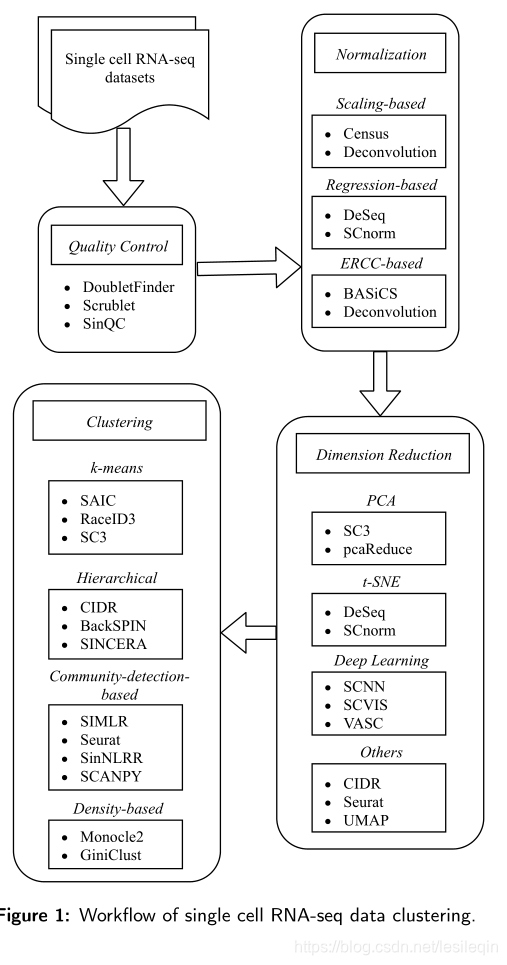
2. Data preprocessing 数据预处理
Given the technical variations and noises, data preprocessing is essential for unsupervised cluster analysis on single-cell RNA-seq data. Quality control is performed to remove the low-quality transcriptomic profile due to capture inefficiency; the single-cell RNA-seq reads should be normalized to remove any amplification biase, sample variation, and other technical confounding factors; dimensional reduction is conducted to project the high-dimensional single-cell RNA-seq data into low-dimensional space. Those upstream steps could have substantial impacts on downstream tasks. Therefore, a myriad of tools have been developed to address the above issues.
考虑到技术差异和噪声,数据预处理对于单细胞RNA-seq数据的无监督聚类分析至关重要。进行质量控制,以消除由于捕获效率低而导致的低质量转录谱;单细胞核糖核酸序列的读数应该正常化,以避免扩增酶、样品变异和其他技术混杂因素;进行降维,将高维单细胞RNA-seq数据投影到低维空间。这些上游步骤可能会对下游任务产生重大影响。 因此,已经开发了无数的工具来解决上述问题。
质量控制 --> 消除捕获效率低而导致的低质量的转录谱
正常化 --> 避免扩增酶、样品变异和其他技术混杂的因素
降维 --> 将高微单细胞RNA-seq数据投影到低维空间
2.1 Quality control 质量控制
Quality control (QC) aims to removing the unreliable cells or genes and other possible missing values for downstream interpretatio. The technical reason for the presence of a large number of cells/genes can be attributed to the doublets with two or more cells suspended in one droplet; on the contrary, a small number of transcripts/genes may result from capture inefficiency (e.g., cell death, cell breakage, and a high fraction of mitochondrial counts) . In this section, we review several stateof-the-art tools or methods in assessing the raw reads and expression matrices of single-cell RNA-seq data. DoubletFinder identifies doublets using gene expression features that significantly improves differential expression analysis performance. Scrublet avoids the need for expert knowledge or cell clustering by simulating multiplets from the data and building a nearest neighbor classifier. SinQC enables us to detect poor data quality, e.g. low mapped reads, a high fraction of mitochondrial counts or low library complexity.
质量控制(QC)旨在去除不可靠的细胞或基因以及其他可能的缺失值,以供下游解释。大量细胞或基因存在的技术原因可归因于两个或更多细胞悬浮在一个液滴中的双重体;相反,少量转录物或基因可能源于捕获效率低下(例如,细胞死亡、细胞断裂和线粒体计数比例高)。在本节中,我们回顾了几种评估单细胞RNA-序列数据的原始读数和表达矩阵的最先进的工具或方法。DoubletFinder利用基因表达特征鉴定了双重基因,显著提高了差异表达分析的性能。通过从数据中模拟多重态并构建最近邻分类器,Scrublet避免了对专家知识或细胞聚类的需要。
2.2 Normalization 正常化
Technical artifacts or experimental noises (e.g. batch effect, insufficient counts,and zero inflation) of high-throughput transcriptomic sequencing may result in differences in expression measurements between samples (e.g. genes). Several studies have revealed that those obvious differences can have a large impact on clustering . Therefore, normalization is essential for adjusting the differences in expression levels across different samples, replicates, or even batches.
高通量转录组测序的技术假象或实验噪音(如批次效应、计数不足和零膨胀)可能导致样本(如基因)之间表达测量的差异。几项研究表明,这些明显的差异会对聚类产生很大影响。因此,标准化对于调整不同样品、重复甚至批次之间表达水平的差异至关重要。
The state-of-the-arts normalization methods have been developed for addressing those issues. We review three kinds of normalization methods as follow: 1) Scaling methods. Lun et al. proposed a strategy to normalize single-cell RNA-seq data with zero counts. Census converts conventional per-cell measures of relative expression values to transcript counts without the need for any spike-in standard or unique molecular identifiers, eliminating much of the apparent technical variability in single-cell experiments; 2) Regression-based methods. DESeq proposed by Anders and Huber adopts local regression to link the variance and mean of negative binomial distribution over the observed counts, resulting in balanced differentially expressed genes. SCnorm uses quantile regression to estimate the dependence of transcript expression on sequencing depth and scale factors to provide normalized expression estimates;3) Methods based on spike-in External RNA Control Cortium (ERCC). Ding et al. presented a normalization tool to remove technical noises and compute for the true gene expression levels based on spike-in ERCC. BASiCS can identify and remove the high and low levels of technical noises (counts).In addition to the above methods, the very simple and commonly used method is to transform read counts using logarithm with a pseudocount such as one.
为解决这些问题,开发了最先进的标准化方法。我们回顾了以下三种归一化(等价于正常化、序列化、标准化)方法:
1)缩放方法。伦等人提出了一种用零计数标准化单细胞RNA-seq数据的策略。Census将传统的每细胞相对表达值的测量方法转换为转录本计数,而不需要任何标准或独特的分子标识符,消除了单细胞实验中的许多明显的技术可变性;
2)基于回归的方法。Anders和Huber提出的DESeq采用局部回归将负二项分布的方差和均值与观察到的计数联系起来,从而产生平衡的差异表达边缘。SCnorm使用分位数回归来估计转录表达对测序深度和比例因子的依赖性,以提供标准化的表达估计;
3)基于棘突内外部RNA控制皮层(ERCC)的方法。丁等人提出了一个归一化工具,以消除技术噪声和计算真正的基因表达水平的基础上,穗在。基础知识(Vallejos等人,2015年)可以识别和消除高水平和低水平的技术噪声(计数)。
除了上述方法之外,非常简单和常用的方法是使用诸如1的伪计数的对数来转换读取计数。
However, those normalization methods also suffer from limitations caused by the diverse assumptions and experimental protocols. The scaling methods cannot account for individual batch effects; the regression-based methods are sensitive to batch effects; ERCC-based methods are not suitable for endogenous and spiked-in transcripts .
然而,这些标准化方法也受到各种假设和实验协议的限制。缩放方法不能考虑单个批次的影响;基于回归的方法对批次效应敏感;基于ERCC的方法不适用于内源性和加标转录物。
2.3 Dimension reduction 降维
Recent advances in single-cell RNA-seq have contributed to measure large-scale expression datasets with hundreds of thousands of genes while it also brings both opportunities and challenges in data analysis. Such high-dimensional gene expression data is unprecedentedly rich and should be well-explored. However, the past clustering methods may be unable to process and interpret such large-scale data. Hence, it is necessary to project the high-dimensional data S. Zhang et al. Page 2 of 12 Review of Single-cell RNA-seq Data Clustering to a lower-dimensional space using dimension reduction that can improve and refine the clustering results. In this section, we review several commonly used dimension reduction methods including principal component analysis, tdistributed stochastic neighbor embedding algorithm, deep learning models, and others.
单细胞RNA-seq的最新进展有助于测量数十万个基因的大规模表达数据集,同时也给数据分析带来了机遇和挑战。这样的高维基因表达数据空前丰富,应该好好发掘。然而,过去的聚类方法可能无法处理和解释这样的大规模数据。因此,有必要利用降维技术将高维数据投影到更低维的空间,以改进和细化聚类结果。在本节中,我们回顾了几种常用的降维方法,包括主成分分析、分布式随机邻居嵌入算法、深度学习模型等。
2.3.1 PCA 主成分分析
Principal Component Analysis (PCA) is a typical linear projection method that projects a set of possibly correlated variables into a set of linearly orthogonal variables (principal components). Due to its conceptual simplicity and efficiency, PCA has been widely used in single-cell RNAseq processing. Notably, SC3 (Kiselev et al., 2017) applied PCA to transform the distance matrices as the input of consensus clustering; Shalek et al. (2014) used PCA for single-cell RNA-seq data spanning several experimental conditions. In addition, some extended and improved PCA-based methods have been developed including pcaReduce (zurauskiene and Yau, 2016) which applied PCA iteratively to provide low-dimensional principal component representations; Usoskin et al. (2015) proposed an unbiased iterative PCA-based process to identify distinct large-scale expression data patterns. However, PCA cannot capture the nonliner relationships between cells because of the high levels of dropout and noise (Kiselev et al., 2019).
主成分分析是一种典型的线性投影方法,它将一组可能相关的变量投影到一组线性正交的变量(主成分)中。由于其概念上的简单性和高效性,主成分分析已被广泛用于单细胞RNA序列处理。值得注意的是,SC3(基舍列夫等人,2017年)应用主成分分析来转换距离矩阵,作为共识聚类的输入;Shalek等人(2014年)将主成分分析用于跨越多个实验条件的单细胞RNA-seq数据。此外,还开发了一些扩展的和改进的基于主成分分析的方法,包括pcaReduce (zurauskiene和Yau,2016),该方法迭代地应用主成分分析来提供低维主成分表示;Usoskin等人(2015)提出了一种基于无偏迭代主成分分析的过程来识别不同的大规模表达数据模式。然而,主成分分析无法捕捉细胞之间的非线性关系,因为脱落和噪声水平高。
2.3.2 t-SNE t分布式随机邻居嵌入
t-distributed Stochastic Neighbor Embedding (t-SNE) is the most commonly used nonlinear dimension reduction method which can uncover the relationships between cells. t-SNE converts data point similarity into probability and minimizes Kullback-Leibler divergence by gradient descent until convergence. In single-cell RNA-seq data analysis, tSNE has become a cornerstone of dimension reduction and visualization for high-dimensional single-cell RNA-seq data. Especially, Linderman et al. (2019) developed a fast interpolation-based t-SNE that dramatically accelerates the processing and visualization of rare cell populations for large datasets. Nonetheless, the limitations of t-SNE include the loss function is non-convex which can lead to different local optimality; the parameters in t-SNE are required to be tuned.
t-分布式随机邻居嵌入(t-SNE)是最常用的非线性降维方法,可以揭示细胞之间的关系。t-SNE将数据点的相似性转化为概率,并通过梯度下降使库勒贝克-莱布勒散度最小化,直到收敛。 在单细胞RNA-seq数据分析中,t-SNE已经成为高维单细胞RNA-seq数据降维和可视化的基石。特别是,林德曼等人(2019年)开发了一种基于快速插值的t-SNE方法,大大加快了大型数据集稀有细胞群的处理和可视化。然而,t-SNE的局限性包括损失函数是非凸的,这可能导致不同的局部最优性;需要调整t-SNE中的参数。
2.3.3 Deep learning models 深度学习模式
In recent years, deep learning models (neural networks and variational auto-encoders) have shown superior performance in interpenetrating complex high-dimensional data. SCNN (Lin et al., 2017a) tested various neural networks architectures and incorporated prior biological knowledge to obtain the reduced dimension representation of single cell expression data. SCVIS (Ding et al., 2018) and VASC (Wang and Gu, 2018) are both based on variational autoencoders which can capture nonlinear relationships between cells and visualize the low-dimensional embedding in singlecell gene expression data.Up to now, those methods demonstrated superior ability of interpretation and compatibility on high-dimensional single-cell RNA-seq data.
近年来,深度学习模型(神经网络和可变自动编码器)在互穿复杂高维数据中显示出优异的性能。SCNN (林等人,2017a)测试了各种神经网络架构,并结合了先前的生物学知识,以获得单细胞表达数据的降维表示。SCVIS(丁等,2018)和VASC(王和顾,2018)都是基于变分自动编码器,该编码器能够捕捉细胞之间的非线性关系并可视化单细胞基因表达数据中的低维嵌入。到目前为止,这些方法在高维单细胞RNA-seq数据上显示出优越的解释能力和兼容性。
2.3.4 Other methods 其他方法
In addition, there are also other dimensional reduction methods such as CIDR (Lin et al., 2017b) applied principal coordinate analysis that preserves the distance information in low-dimension space from its high-dimension space; Seurat (Butler et al., 2018) is a toolkit for analysis of single cell RNA sequencing data and provides many dimension reduction methods such as PCA and t-SNE. Uniform Manifold Approximation and Projection (UMAP) (Mcinnes et al., 2018) is a widely used technique for dimension reduction. UMAP provides increased speed and better preservation of data global structure for high dimensional datasets. It has been verified that it outperforms t-SNE (Becht et al., 2019).
此外,还有其他降维方法,如应用主坐标分析的(林等,2017b),该方法从高维空间保留低维空间中的距离信息;修拉(Butler等人,2018)是一个用于分析单细胞RNA测序数据的工具包,提供了许多降维方法,如PCA和t-SNE。均匀流形近似和投影(UMAP)(麦金尼斯等人,2018)是一种广泛使用的降维技术。UMAP为高维数据集提供了更快的速度和更好的数据全局结构保存。已经证实它优于t-SNE (Becht等人,2019年)。
3. Clustering methods for single-cell RNA-seq 单细胞RNA-seq测序的聚类方法
Diverse types of clustering methods have been developed for detecting cell types from single-cell RNA-seq data. Those methods can be roughly classified into four categories including k-means clustering, hierarchical clustering, community-detection-based clustering, and density-based clustering. We review several computational applications of those clustering methods with their strengths and limitations. Table1illustrates the overview of the state-of-the-arts clustering methods on single-cell RNA-seq data.
已经开发了不同类型的聚类方法来从单细胞RNA-序列数据中检测细胞类型。这些方法可以大致分为四类,包括k-均值聚类、层次聚类、基于社区检测的聚类和基于密度的聚类。我们回顾了这些聚类方法的几种计算应用及其优势和局限性。下表展示了单细胞RNA-序列数据的最先进的聚类方法的概述。

3.1 k-means clustering k均值聚类
k-means clustering is the most popular clustering approach, which iteratively finds a predefined number of k cluster centers (centroids) by minimizing the sum of the squared Euclidean distance between each cell and its closest centroid. In addition, it is suitable for large datasets since it can scale linearly with the number of data points (Lloyd, 1982).
k-means聚类是最流行的聚类方法,它通过最小化每个像元与其最近质心之间的平方欧几里德距离之和来迭代地找到预定数量的k聚类中心(质心)。此外,它适用于大型数据集,因为它可以随着数据点的数量线性扩展(劳埃德,1982)。
Several clustering tools based on k-means have been developed for interpreting single-cell RNA-seq data. SAIC (Yang et al.,2017) utilized an iterativek-means clustering to identify the optimal subset of signature genes that separate single cells into distinct clusters. pcaReduce (zurauskiene and Yau, 2016) is a hierarchical clustering method while it relies on k-means results as the initial clusters. RaceID (Grün et al., 2015) applied k-means to unravel the heterogeneity of rare intestinal cell types (Tibshirani et al., 2001).
已经开发了几种基于k-means的聚类工具来解释单细胞RNA-seq数据。SAIC 利用iterative k-means聚类来识别将单细胞分成不同簇的特征基因的最佳子集。pcard uce是一种分层聚类方法,它依赖于k-means结果作为初始聚类。RaceID应用k-means揭示了罕见肠细胞类型的异质性。
However, k-means clustering is an greedy algorithm that may fail to find its global optimum; the predefined number of clusters k can affect the clustering results; and another disadvantage is its sensitivity to outliers since it tends to identify globular clusters, resulting in the failures in detecting of rare cell types.
然而,k-means聚类是一种贪婪算法,可能无法找到其全局最优;k聚类的预定数量会影响聚类结果;另一个缺点是它对异常值的敏感性,因为它倾向于识别球状星团,导致检测稀有细胞类型的失败。
To overcome the above drawbacks, SC3 (Kiselev et al., 2017) integrated individual k-means clustering results with different initial conditions as the consensus clusters. RaceID2 (Grün et al., 2016) replaced the k-means clustering with kmedoids clustering that use 1- pearson’s correlation instead of Euclidean distance as the clustering distance metric. RaceID3 (Herman et al., 2018), as the advanced version of RaceID2 added feature selection and introduced random forest to reclassify k-means clustering results.
为了克服上述缺点,SC3 (Kiselev等人,2017年) 将个体k-means聚类结果与不同的分类条件(感知聚类)相结合。RaceID2用k-medoids聚类代替了k-means聚类,k-medoids聚类使用1- pearson相关性而不是欧几里德距离作为聚类距离度量。RaceID3作为RaceID2的高级版本,增加了特征选择并引入了随机森林来重新分类k-means聚类结果。
3.2 Hierarchical clustering 分层聚类
Hierarchical clustering is another widely used clustering algorithm on single-cell RNA-seq data. There are two types of hierarchical strategies including: 1) agglomerative clustering, the individual cells are progressively merged into clusters according to distance measures; 2) divisive clustering, each cluster is split into small groups recursively until individual data level. These two strategies build a hierarchical structure among the cells/genes and enable the improvement in finding rare cell types as small clusters. Hierarchical clustering does not require pre-determining the number of clusters and make assumptions for the distributions of single-cell RNA-seq data. Hence, many single-cell RNA-seq clustering methods have adopted it as part of the computational component.
分层聚类是另一种广泛使用的单细胞RNA-seq数据聚类算法。有两种分层策略,包括:
1)聚集聚类,根据距离度量将单个单元逐步合并成聚类;
2)分裂聚类,每个聚类被递归地分成小组,直到单个数据级别
这两种策略在细胞/基因之间建立了一个层次结构,并能够在发现作为小簇的稀有细胞类型方面有所改进。分层聚类不需要预先确定聚类的数量,也不需要对单细胞RNA-seq数据的分布做假设。因此,许多单细胞RNA-序列聚类方法已经将其作为计算组件的一部分。
CIDR (Lin et al., 2017b) integrates both dimension reduction and clustering based on hierarchical clustering into single-cell RNA-seq analysis and uses implicit imputation process for dropout effects; it provides a stable estimation of pairwise cells distances. BackSPIN developed a biclustering method based on divisive hierarchical clustering and sorting points into neighborhoods (SPIN) (Tsafrir et al., 2005) to simultaneously cluster genes and cells. The number of splits need to be set manually in BackSPIN. Although intensive splits can improve the clustering resolution, it is prone to over-partition. pcaReduce (zurauskiene and Yau, 2016) is an agglomerative hierarchical clustering approach with PCA which provides clustering results in a hierarchical. SINCERA (Guo et al., 2015) as a simple pipeline adopted hierarchical clustering with centered PearsonâĂŹs correlation and average linkage method to identify cell types.
CIDR将基于层次聚类的降维和聚类结合到单细胞RNA-seq分析中,并使用隐式插补过程来处理缺失效应;它提供了成对小区距离的稳定估计。 BackSPIN开发了一种基于划分层次聚类和将点分类到邻域的双聚类方法,以同时聚类基因和细胞。拆分的数量需要在BackSPIN中手动设置。虽然密集分割可以提高聚类分辨率,但容易过度分割。pcaredcuce是一种具有主成分分析的凝聚层次聚类方法,它以层次结构提供聚类结果。SINCERA作为一个简单的流水线,采用了具有中心pearsonâăźs 相关的层次聚类和平均链接方法来识别细胞类型。

聚集层次聚类具有O(N3),而分裂聚类是O(2N).虽然层次聚类可以揭示细胞/基因之间的层次关系,不需要设置聚类数,但时间复杂度较高。
3.3 Community-detection-based clustering 基于社团检测的聚类
Given the limitations of k-means and hierarchical clustering methods in large-scale datasets, community-detectionbased clustering has been increasingly popular recently. Community detection is crucial in sociology, biology, and other systems that can be represented as graphs with nodes and edges. For single-cell RNA-seq data, nodes refer to cells and edge weights are represented by cell-cell pairwise distances.The idea of graph-based clustering is to delete the S. Zhang et al. Page 4 of 12 Review of Single-cell RNA-seq Data Clustering branch with maximum weights (cell-cell pairwise distances) in a dense graph (cell relationship network). There are three commonly used approaches for community-detectionbased (graph-based) clustering including clique algorithm, spectral clustering, and Louvain algorithm (Blondel et al., 2008).
鉴于k-means聚类和层次聚类方法在大规模数据集上的局限性,基于社区检测的聚类近年来越来越受欢迎。社区检测在社会学、生物学和其他可以用带节点和边的图表示的系统中至关重要。对于单细胞RNA-seq数据,节点指的是细胞,边缘权重由细胞间的成对距离表示。基于图的聚类的思想是在密集图(细胞关系网络)中删除具有最大权重(细胞-细胞成对距离)的单细胞RNA-序列数据聚类分支。基于社区检测(基于图)的聚类有三种常用方法,包括团算法、谱聚类和卢万算法。
A clique is a set of points fully connected to each other in a graph and represents a cluster (community). Although finding cliques in a graph is NP-complete, some studies have been conducted to address it such as heuristic optimization. SNN-Clip (Xu and Su, 2015) was proposed to leverage the concept of shared nearest neighbor to calculate cell similarity (Zhang et al., 2009) for finding all quasi-cliques since the shared nearest neighbor graph is sparse. SNN-Clip does not require specifying the number of clusters manually while it is non-scalable and the resultant clusters are not stable.
团是在一个图中相互完全连接的一组点,代表一个簇(群体)。虽然在图中寻找团是NP完全的,但是已经有一些研究来解决这个问题,例如启发式优化。SNN-Clip提出利用共享最近邻的概念来计算细胞相似性以发现所有的准集团,因为共享最近邻图是稀疏的。SNN-Clip不需要手动指定群集的数量,因为它是不可扩展的,并且生成的群集不稳定。
Spectral clustering is a widely used clustering method recently. It is designed to be adaptive to data distribution by relying on the eigenvalues of cell similarity matrix. Nonetheless, the spectral clustering’s time complexity is O(N3). SIMLR (Wang et al., 2017) is an analytic framework for dimension reduction, clustering, and visualization of single-cell RNA-seq data. It is a method specificially designed at single-cell RNA-seq. SIMLR combines spectral clustering with multiple kernel similarity measures for clustering expression data generated from cross-platform and cross-condition experiments. In addition, SIMLR has an advantage in processing large-scale datasets with heavy noises. SinNLRR (Zheng et al., 2019) was proposed to impose a non-negative and low rank structure on cell similarity matrix and then applied spectral clustering to detect cell types. However, the spectral clustering requires users to set the number of clusters in the data.
谱聚类是近年来广泛使用的聚类方法。它被设计成通过依赖单元相似性矩阵的特征值来适应数据分布。尽管如此,光谱聚类的时间复杂度是O(N3).SIMLR是用于单细胞RNA-seq数据的降维、聚类和可视化的分析框架。这是一种专为单细胞RNA-序列设计的方法。SIMLR将谱聚类与多核相似性度量相结合,用于对跨平台和跨条件实验生成的表达式数据进行聚类。另外,SIMLR在处理噪声较大的大规模数据集时具有优势。SinNLRR提出将**非负低秩结构强加于细胞相似性矩阵,然后应用谱聚类来检测细胞类型。**然而,谱聚类要求用户设置数据中的聚类数。
Louvain (Blondel et al., 2008) is the most popular community detection algorithm and widely used to single-cell RNA-seq data. The time complexity of Louvain is O(N log(N)) which is lower than other community-detection-based algorithms. SCANPY (Wolf et al., 2018) is a scalable toolkit for single-cell RNA-seq analysis and its clustering section is based on Louvain algorithm. SCANPY has advantages in scaling its computation with the number of cells (over one million). Seurat (Satija et al., 2015) also applied Louvain algorithm to cluster the cell types for the mapping of cellular localization.
Louvain 是最流行的社区检测算法,广泛用于单细胞RNA-seq数据。Louvain 的时间复杂度是O(N log(N)),其低于其他基于社区检测的算法。SCANPY是一个可扩展的单细胞RNA-seq分析工具包,其聚类部分基于Louvain算法。SCANPY在根据单元数量(超过一百万)缩放计算方面具有优势。Seurat 还应用Louvain算法对细胞类型进行聚类,以绘制细胞定位图。
3.4 Density-based clustering 基于密度的聚类
Density-based clustering methods separate data space into highly dense clusters. It can learn clusters with arbitrary shapes and identify noises (outliers). The most popular density-based clustering algorithm is DBSCAN (Ester et al., 1996). DBSCAN does not need to predetermine the number of clusters and its time complexity is O(N log(N)). However, DBSCAN requires user to set two parameters including э (eps) and the minimum number of points required to form a dense region (minPts) (Ester et al., 1996) that will affect its clustering results. Jiang et al. (2016a) developed GiniClust, detecting rare cell types from single-cell gene expression data and its clustering method is based on DBSCAN. GiniClust is effective in finding rare cell types since it can be adaptively adjusted to set a lower э. However, such a design may lead to unreasonable large cell clusters. Monocle2 (Qiu et al., 2017b) also applied DBSCAN to identify the differential expressed genes between cells.
基于密度的聚类方法将数据空间分成高密度的聚类。它可以学习任意形状的聚类,识别噪声(离群值)。最流行的基于密度的聚类算法是DBSCAN。DBSCAN不需要预先确定集群的数量,其时间复杂度为O(N log(N)).然而,DBSCAN要求用户设置两个参数,包括 э (eps)和形成密集区域(minPts)所需的最小点数,这将影响其聚类结果。蒋等提出了基于单个细胞基因表达数据检测稀有细胞类型的Gini cluster,其聚类方法基于DBSCAN。giniclust在发现罕见细胞类型方面是有效的,因为它可以被适应性地调整以设置较低的э。然而,这样的设计可能导致不合理的大细胞群。单克隆抗体2(邱等,2017b)也应用DBSCAN来鉴定细胞间差异表达的基因。
4. Density-based clustering 聚类方法的实验评估
In this section, we conduct independent experiments to evaluate several widely used single-cell RNA-seq clustering methods. Those clustering methods contain RaceID3 (Herman et al., 2018), Monocle2 (Qiu et al., 2017b), SIMLR (Wang et al.,2017), Seurat (Satija et al.,2015), SC3 (Kiselev et al., 2017), and CIDR (Lin et al., 2017b). We applied six single-cell RNA-seq clustering methods on two different droplet-based transcriptomic datasets (GSE84133 and GSE65525) with cell types annotations. For the evaluation and comparison, we introduce two commonly used metrics including Adjusted Rand index, Running Time, and Homogeneity Score to measure the clustering performance and efficiency respectively. The parameter setting of the cluster methods on both datasets are tabulated in Table 2. In particular, we would like to note that most of parameters were chosen based on the default setting given by individual methods.
在这一部分,我们进行独立的实验来评估几种广泛使用的单细胞RNA-序列聚类方法。这些聚类方法包括RaceID3 、Monocle2、SIMLR、Seurat、SC3 和 CIDR。我们在两个不同的基于液滴的带有细胞类型注释的转录组数据集(GSE84133和GSE65525)上应用了六种单细胞RNA-seq聚类方法。为了评估和比较,我们引入了两个常用的指标,包括调整兰德指数、运行时间和同质性分数,分别用来衡量聚类的性能和效率。 下表列出了两个数据集上聚类方法的参数设置。特别是,我们要注意的是,大多数参数都是根据单个方法给出的默认设置选择的。

4.1 Evaluation metrics for clustering 聚类评估指标
Since the single-cell RNA-seq clustering is an unsupervised learning task in most studies, three common metrics Adjusted Rand index, Running Time, and Homogeneity Score are introduced for the evaluation.
由于单细胞核糖核酸序列聚类在大多数研究中是一项无监督的学习任务,因此引入了三个常用指标:校正兰德指数、运行时间和同质性分数来进行评估。
ARI校正兰德指数:
Adjusted Rand index (ARI) proposed by Hubert and Arabie(1985) can be used to measure the similarity between the clustering results of interest and the true clustering. However, ARI is widely applied as the metric of single-cell RNA-seq clustering only when the cell-labels are available (Kiselev et al., 2017; Ntranos et al., 2016; Lin et al., 2017b; Aibar et al., 2017; Xu and Su, 2015). Given a set of n cells and two clusterings (X = {X1, X2, …, Xs} partitioned by clustering method and Y = {Y1, Y2, …, Yr} partitioned by annotated cell types) of these cells, the overlap between the two clusterings can be summarized in a contingency table with s rows and r columns. The ARI is defined as below.
休伯特和阿拉比提出的调整兰德指数(ARI)可以用来衡量感兴趣的聚类结果和真实聚类之间的相似性。然而,只有当细胞标记可用时,ARI才被广泛用作单细胞RNA-序列聚类的度量。给定一组n单元和两个集群 (X = {X1,X2,…,Xs} 用聚类法分区,Y = {Y1,Y2,…Yr}由带注释的单元类型划分),这两个集群之间的重叠可以在具有s行和r列的列联表中总结。ARI的定义如下。
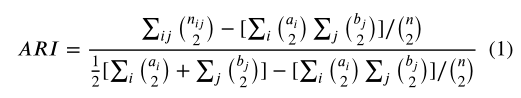
where n i j = ∣ X i ∩ Y j ∣ n_{ij} = | X_i \cap Y_j | nij=∣Xi∩Yj∣ denotes the values from the contingency table; a i = ∑ j n i j a_i=\sum_jn_{ij} ai=∑jnij and b j = ∑ i n i j b_j=\sum_in_{ij} bj=∑inij represent the i i ith row sums and j j jth column sums of the contingency table, respectively; and () denotes a binomial coefficient. ARI = 1 indicates a perfect overlap between clusters X and Y , while ARI = 0 indicates random clustering.
其中 n i j = ∣ X i ∩ Y j ∣ n_{ij} = | X_i \cap Y_j | nij=∣Xi∩Yj∣表示列联表中的值; a i = ∑ j n i j a_i=\sum_jn_{ij} ai=∑jnij 和 b j = ∑ i n i j b_j=\sum_in_{ij} bj=∑inij分别表示列联表的 i i ith行和与 j j jth列和;() 表示二项式系数。ARI = 1表示集群X和Y之间完全重叠,而ARI = 0表示随机集群。
Homogeneity Score同质性得分
Homogeneity Score (Rosenberg and Hirschberg, 2007) evaluates the performance of clustering results with regards to the ground truth.It is defined as:
同质性得分评估了聚类结果与基本事实的关系。它的定义如下:
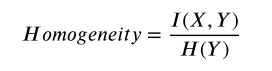
where H ( Y ) = I ( Y , Y ) H(Y) = I(Y ,Y) H(Y)=I(Y,Y) is the entropy of Y Y Y and I ( X , Y ) I(X,Y) I(X,Y) is the mutual information of X X X and Y Y Y. It is bounded between 0 and 1. Homogeneity = 1 indicates all of its clusters contain only data points from a single class while low values indicate that clusters contain mixed known groups.
其中 H ( Y ) = I ( Y , Y ) H(Y) = I(Y ,Y) H(Y)=I(Y,Y)是 Y Y Y 和 I ( X , Y ) I(X,Y) I(X,Y)是 X X X和 Y Y Y的互信息它在0和1之间有界。同质性= 1表示它的所有聚类只包含来自单个类的数据点,而低值表示聚类包含混合的已知组。
Running time运行时间:
In addition, running time is usually measured to evaluate the algorithm efficiency. High efficiency is an important feature since the single-cell RNA-seq data usually come up with thousands of cells and genes.
此外,通常测量运行时间来评估算法效率。高效率是一个重要的特征,因为单细胞RNA-seq数据通常会产生数千个细胞和基因。
4.2 Performance on mouse pancreas single-cell RNA-seq dataset (GSE84133) 在小鼠胰腺单细胞核糖核酸数据集上的表现
In mouse pancreas single-cell RNA-seq dataset (GSE84133) (Baron et al.,2016), there are 1,886 cells in 13 cell types after the exclusion of hybrid cells.GSE84133 has 14,878 genes.Figure 2 and 3 shows the ARI, homogeneity scores, and running time of RaceID3, Monocle2, SIMLR, Seurat, SC3, and CIDR on GSE84133 for performance comparision.
在小鼠胰腺单细胞RNA-seq数据集(GSE84133)中,排除杂交细胞后,在13种细胞类型中有1886个细胞。GSE84133有14878个基因。下面的图显示了GSE84133上RaceID3、Monocle2、SIMLR、Seurat、SC3和CIDR的ARI、同质性分数和运行时间,以进行性能比较。
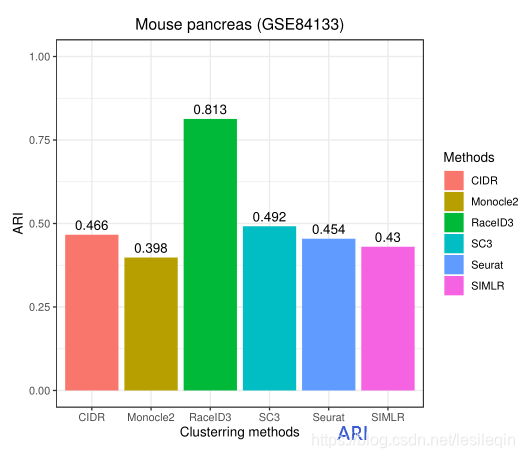
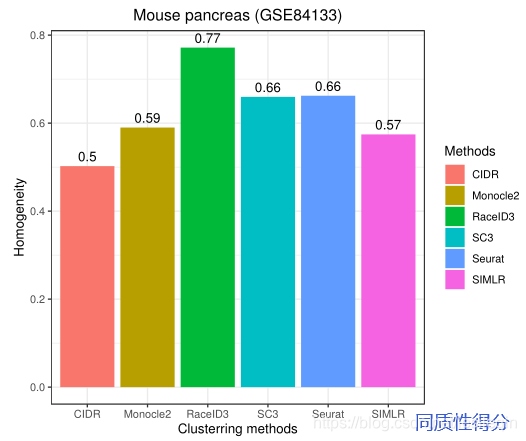
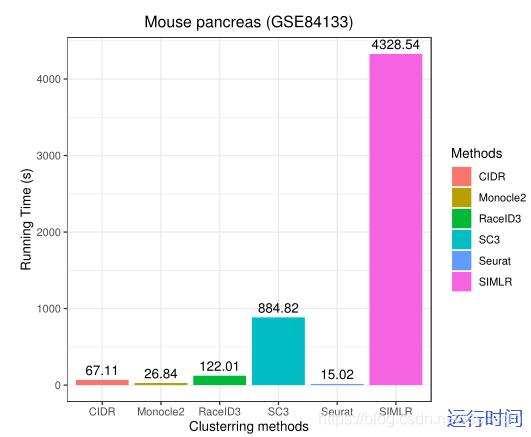
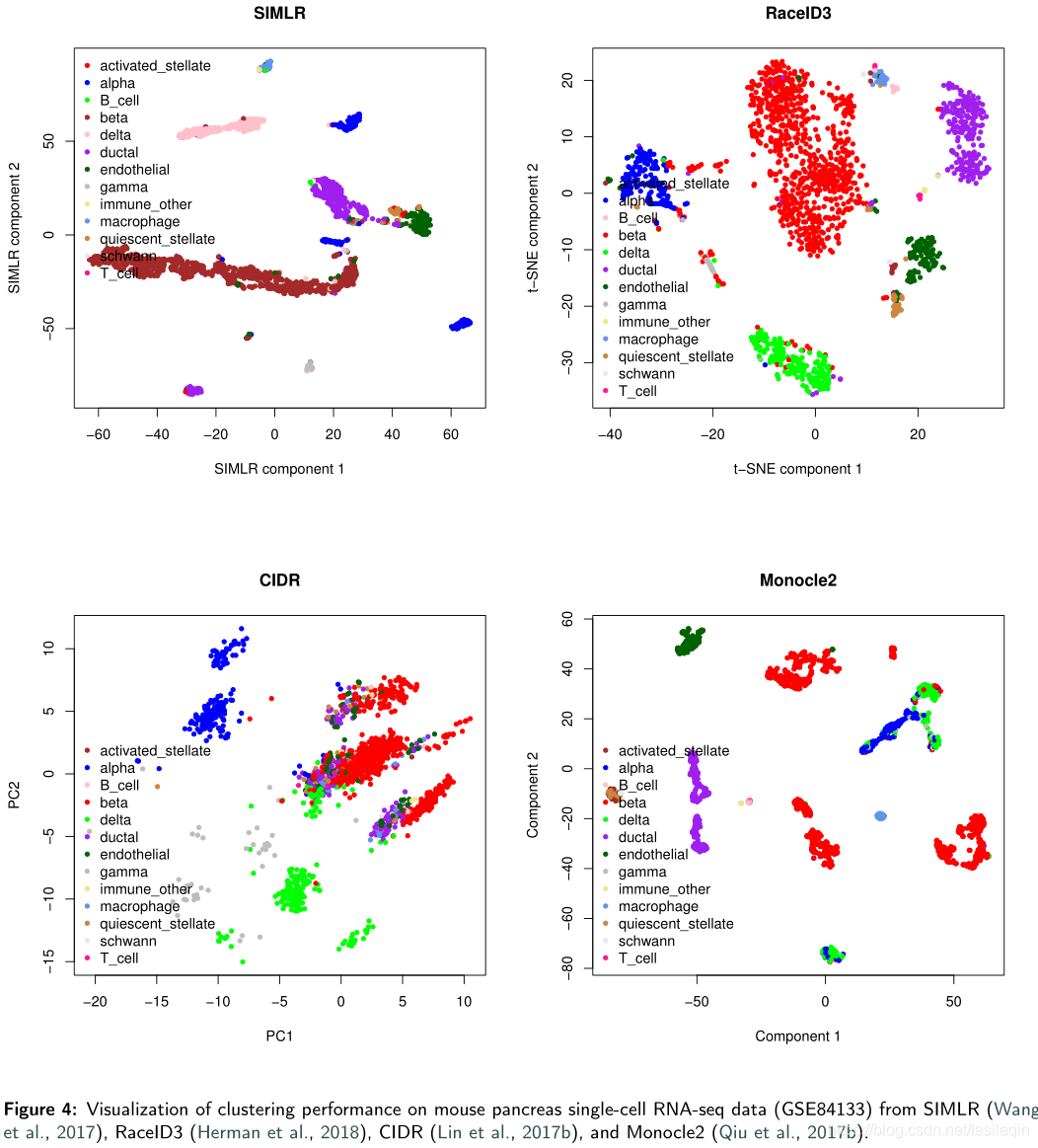
The results show that RacID3 exhibit the best ARI (=0.813) and homogeneity (=0.77) performance among the six methods.The ARI of other methods does not exceed 0.500. SIMLR is a time-consuming method and it took 1.20 hours to conduct the clustering task.However, CIDR can only identify seven cell types from GSE84133 since it belongs to hierarchical clustering and is unable to predetermine the number of clusters. SC3, SIMLR, and Monocle2 cannot provide an accurate estimation of the cluster count and it has to be determined manually. Seurat, Monocle2, and RaceID3 require user to adjust multiple parameters to achieve the best clustering performance that affected the user friendliness.Figure 4 illustrates the clustering performance of SIMLR, RaceID3, CIDR, and Monocle2 on GSE84133.
结果表明,在六种方法中,RacID3表现出最好的ARI (=0.813)和同质性(=0.77)。其他方法的ARI不超过0.500。SIMLR是一种耗时的方法,执行聚类任务需要1.20个小时。然而,CIDR只能从GSE84133中识别七种细胞类型,因为它属于分级聚类,并且不能预先确定聚类的数量。SC3、SIMLR和Monocle2不能提供群集计数的准确估计,必须手动确定。Seurat, Monocle2, and RaceID3 要求用户调整多个参数以获得影响用户友好性的最佳聚类性能。下图展示了SIMLR、RaceID3、CIDR和Monocle2在GSE84133上的集群性能。
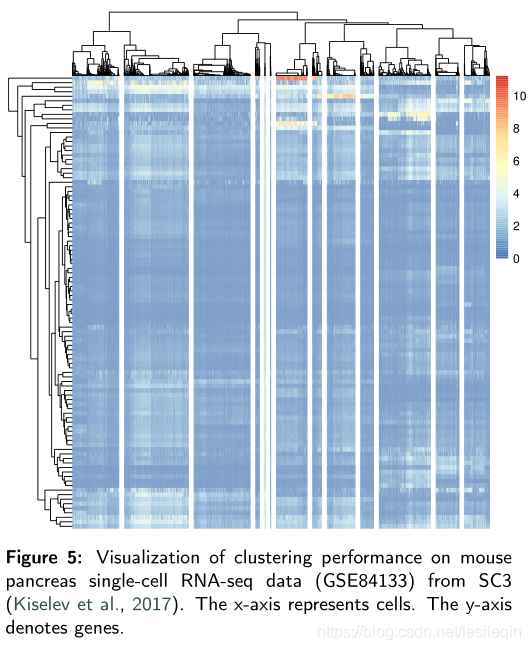
The visualization results are directly obtained from their R packages. Figure 5 displays the clustering results from SC3. Since SC3 belongs to hierarchical clustering, the clustering result is illustrated in heatmap and it is set to show the 13 cell types.
可视化结果直接从他们的R包中获得。下图显示了来自SC3的聚类结果。由于SC3属于分层聚类,聚类结果显示在热图中,并设置为显示13种单元类型。
4.3 Performance on mouse embryonic stem single-cell RNA-seq dataset (GSE65525) 小鼠胚胎干细胞单细胞核糖核酸序列数据集的性能
In mouse embryonic stem single-cell RNA-seq largescale dataset (GSE65525) (Klein et al., 2015), there are 2717 cells in four annotated cell types. GSE65525有24,175个基因。Figure 6 and 7 depict the ARI, homogeneity scores, and running time of RaceID3, Monocle2, SIMLR, Seurat, SC3, and CIDR on GSE65525 for performance comparison.
在小鼠胚胎干细胞单细胞RNA-seq大规模数据集(GSE65525) (Klein等人,2015)中,在四种注释的细胞类型中有2717个细胞。GSE65525有24175个基因。下面的图描述了GSE65525上的RaceID3、Monocle2、SIMLR、Seurat、SC3和CIDR的ARI、同质性分数和运行时间,以进行性能比较。
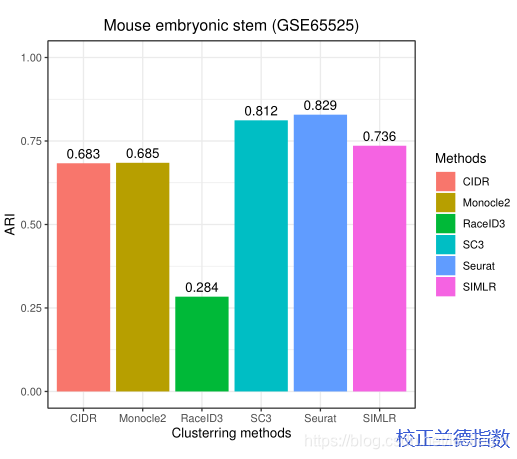
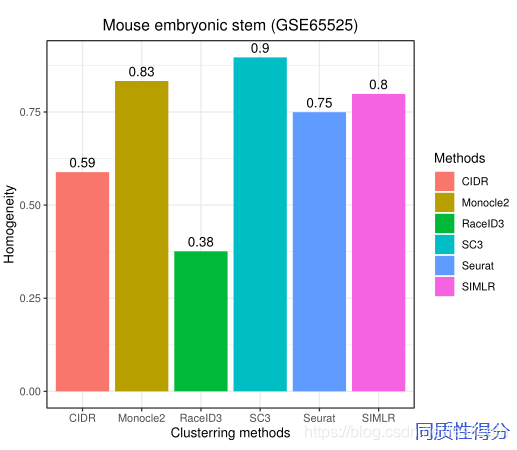
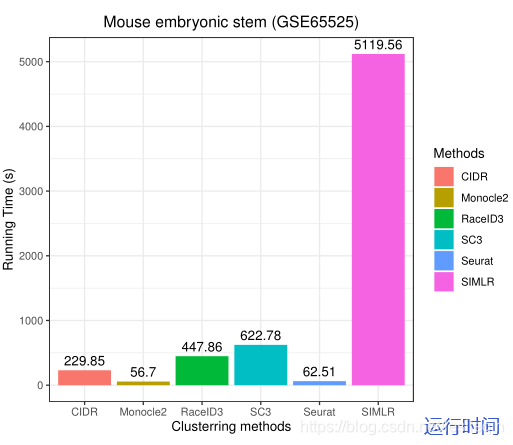
The results show that exhibits the best ARI (=0.829) performance among the six methods, while its homogeneity score (homogeneity=0.75) is lower than SC3 (homogeneity=0.9).The ARI of SC3 (0.812) also exceeds 0.80 and it achieve higher homogeneity than others.Hence, SC3 exhibits robust clustering performance on large datasets with a reasonable sacrifice on efficiency. The ARI and homogeneity scores of Monocle2, SIMLR, RaceID3, SC3, and CIDR showed different degrees of accuracy. RaceID achieved the worst ARI (=0.284) and homogeneity score (homogeneity=0.38) across six methods on GSE65525. SIMLR has been run for 85 minutes which took far more than other five methods.Hence, the results show that RaceID3 may not be suitable to large-scale singlecell RNA-seq datasets.Figure 8 illustrates the clustering performance of SIMLR, RaceID, CIDR, and Monocle2 on GSE65525.
结果表明,在六种方法中,Seurat 的ARI = 0.829最好,而其同质性得分(同质性=0.75)低于SC3(同质性=0.9)。SC3的ARI(0.812)也超过了0.80,它达到了比其他更高的同质性。因此,SC3在大数据集上表现出稳健的聚类性能,但在效率上有合理的牺牲。Monocle2、SIMLR、RaceID3、SC3和CIDR在ARI和同质性得分上展示了不同程度的准确性。RaceID在GSE65525的六种方法中获得了最差的ARI (=0.284)和同质性得分(同质性=0.38)。SIMLR 运行了85分钟,远远超过了其他五种方法。因此,结果表明RaceID3可能不适合大规模的单细胞RNA-seq数据集。下图展示了SIMLR、RaceID、CIDR和Monocle2在GSE65525上的集群性能。
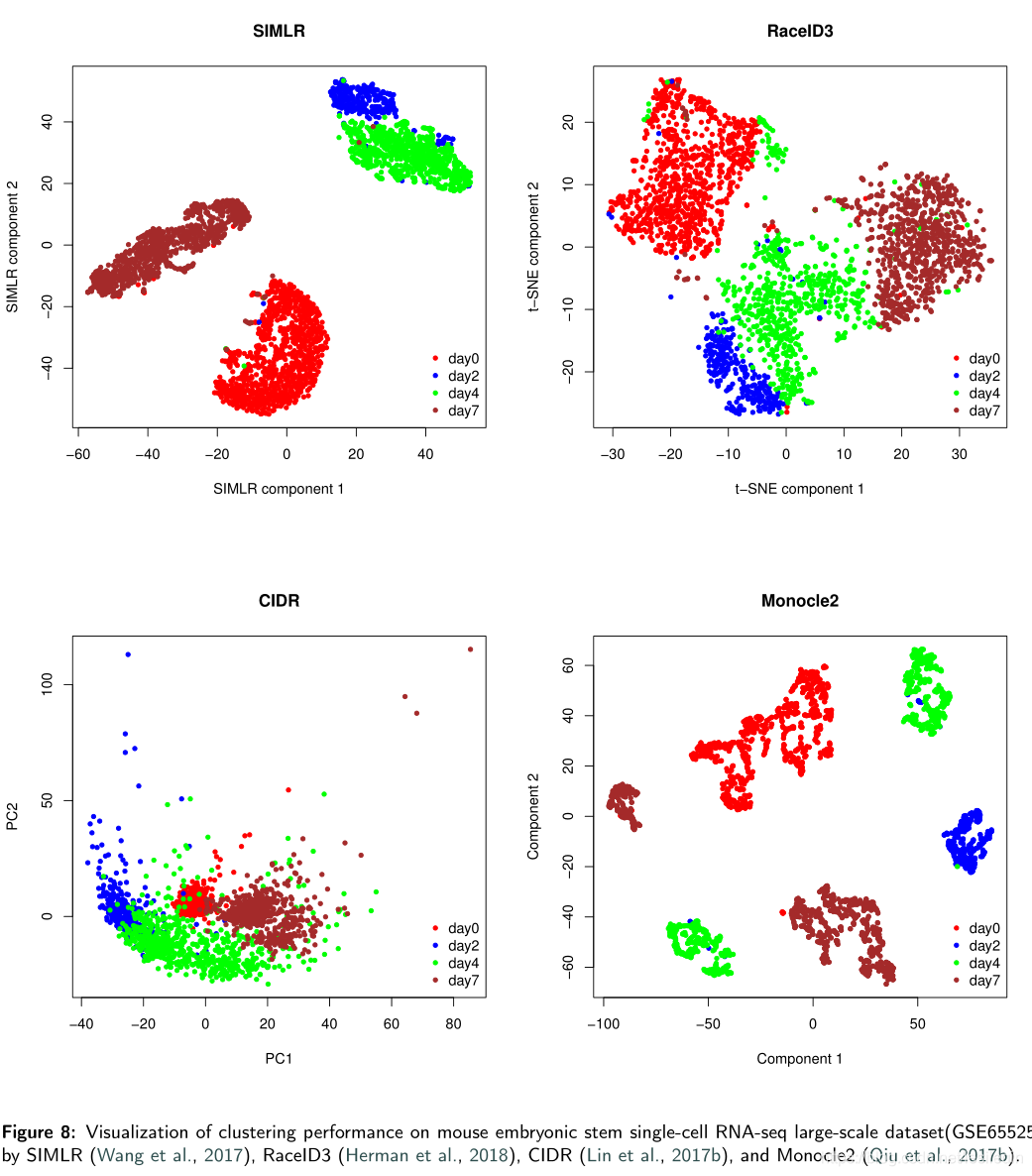
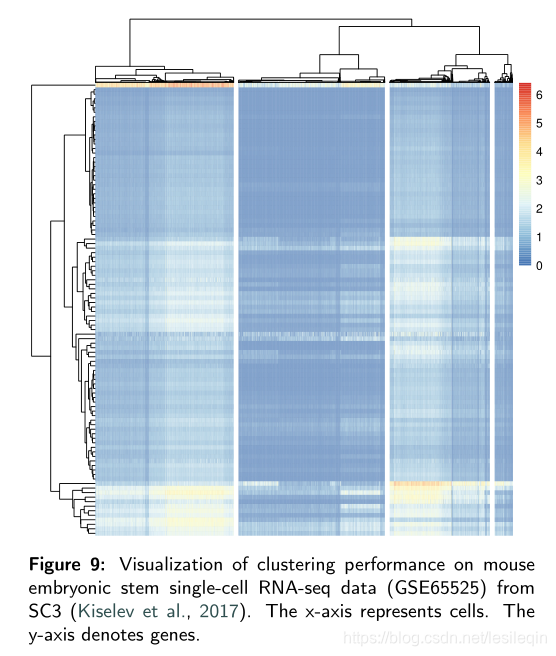
The visualization results are directly obtained from their R packages. Results from Figure 8 show that all methods result in different degrees of undesirable overlaps between clusters. Figure 9 displays the clustering results of SC3 on GSE65525 and it shows the four correct cell types.
可视化结果直接从他们的R包中获得。上图的结果显示,所有方法都会导致集群之间不同程度的不良重叠。下图显示了SC3在GSE65525上的聚类结果,并显示了四种正确的单元格类型。
5. Discussions and conclusions 讨论和结论
Single-cell RNA-seq data analysis is a crucial component in whole-transcriptome studies. In particular, data clustering is the central component of single-cell RNA-seq analysis.Clustering results can affect the performance of downstream analysis including identifying rare or new cell types, gene expression patterns that are predictive of cellular states, and functional implications of stochastic transcription.
单细胞RNA-序列数据分析是全转录组研究的重要组成部分。特别是,数据聚类是单细胞RNA-seq分析的核心组成部分。聚类结果会影响下游分析的性能,包括识别稀有或新的细胞类型、预测细胞状态的基因表达模式以及随机转录的功能含义。
There are several related studies for the performance evaluation of clustering methods on single-cell RNA-seq data.Those studies focused on assessing the methods for clustering single-cell RNA-seq data, while the data preprocessing steps may not be included in the respective discussion section, although it could have significantly influences on the downstream clustering performance.Therefore, in this study, we reviewed several clustering methods.In addition, the upstream RNAseq data preprocessing steps have also been reviewed since those steps can significantly affect the downstream clustering performance. Lastly, our performance comparison experiments have also been conducted, revealing independent insights into the state-of-the-arts methods without any conflict of interest.
对单细胞RNA-seq数据聚类方法的性能评价有几个相关的研究。这些研究侧重于评估单细胞RNA-seq数据的聚类方法,而数据预处理步骤可能不包括在相应的讨论部分,尽管它可能对下游聚类性能产生重大影响。因此,在这项研究中,我们回顾了几个聚类方法。此外,还回顾了上游RNAseq数据预处理步骤,因为这些步骤会显著影响下游聚类性能。最后,我们还进行了性能比较实验,揭示了对最新方法的独立见解,没有任何利益冲突。
Those clustering methods show expected performance on single-cell RNA-seq data. However, those clustering methods have its drawbacks; for instance, k-means clustering require users to determine the number of clusters and is sensitive to outliers; hierarchical clustering has high complexity and may be unsuitable to large-scale singlecell RNA-seq data; community-detection-based clustering cannot provide the estimation of number of clusters and is unsuitable for small communities; density-based clustering has advantages in detecting rare cell types with a sacrifice on large cluster performance.
这些聚类方法显示了单细胞RNA-序列数据的预期性能。然而,这些聚类方法有其缺点;例如,k-均值聚类要求用户确定聚类的数量,并且对异常值敏感;层次聚类具有很高的复杂性,可能不适合大规模的单细胞RNA-seq数据;基于社区检测的聚类不能提供聚类数目的估计,并且不适用于小社区;基于密度的聚类在检测稀有细胞类型方面具有优势,但会牺牲大型聚类的性能。
In addition to those limitations, there are still some technical challenges in single-cell RNA-seq clustering. With the advanced development of single-cell RNA-seq techniques, the single-cell datasets are growing to be extremely highdimensional and sparse. Although some methods can deal with those data in a time span of hours such as SIMLR, visualization of those data is still a challenge. Moreover, the low dimensionality of expression profiles implies intensive gene co-expression signature that may inspire us to develop new clustering methods on low-dimensional data to interpret cell types (Crow and Gillis, 2018). Advanced data integration and analysis approaches are needed for both basic research and clinical studies in the coming years.
除了这些限制之外,单细胞RNA-序列聚类还存在一些技术挑战。随着单细胞RNA-seq技术的发展,单细胞数据集变得非常高维和稀疏。虽然有些方法可以在几个小时内处理这些数据,例如SIMLR,但是这些数据的可视化仍然是一个挑战。此外,表达谱的低维度意味着密集的基因共表达信号,这可能会激励我们在低维度数据上开发新的聚类方法来解释细胞类型(Crow和Gillis,2018)。未来几年,基础研究和临床研究都需要先进的数据集成和分析方法。
- 仅作为个人学习笔记,如果有遇翻译错误,还请在评论区多多指正
- 本文基于
知云文献翻译


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









