






目录
0.注意事项:
1.scf.in,nscf.in,twin.in的晶格信息,赝势信息要一致!!!
2.使用sctk.x时 -np和-nk一定要一致!!!
3.需要nbnd的文件:nscf.in,twin.in,sctk.in请保持一致
4.scf.in nk>k点个数会报错,输出文件CRASH,
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
task # 24
from divide_et_impera : error # 1
some nodes have no k-points
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%

1. 自洽计算——算电子密度
scf.in
&CONTROL
calculation = 'scf'
pseudo_dir = './'
/
&SYSTEM
ibrav = 0
nat = 3
ntyp = 2
ecutwfc = 60.0
ecutrho = 240.0
注意这里一定要用这种方法!
occupations = 'tetrahedra_opt'
/
&ELECTRONS
mixing_beta = 0.3
conv_thr = 3.000000e-10
/
CELL_PARAMETERS angstrom
3.072263267 0.000000000 0.000000000
-1.536131635 2.660657665 0.000000000
0.000000000 0.000000000 3.511486466
ATOMIC_SPECIES
B 10.811000 B.pbe-nc.UPF
Mg 24.305000 Mg.pbe-n-nc.UPF
ATOMIC_POSITIONS crystal
Mg 0.000000 0.000000 0.000000
B 0.333333 0.666667 0.500000
B 0.666667 0.333333 0.500000
K_POINTS automatic
8 8 6 0 0 0
export OMP_NUM_THREADS=1 #只允许单线程
$ mpiexec -np 29 PATH/pw.x -nk 29 -in scf.in > scf.out #PATH可执行文件路径
重要的参量:
calculation = “scf”
用pw.x进行自洽计算
注意:我们可以先计算声子和电声相互作用也可以先计算库伦作用。(大概是说第二步和第三步的计算顺序无所谓?)
生成文件:
scf.out
pwscf.save/ #文件夹
pwscf.save/charge-density.dat
pwscf.save/data-file-schema.xml
pwscf.save/wfc*.dat #MgB2一共40个
#是scf.out中
#number of k points= 40 (tetrahedron method)
#这个数量和np,nk无关
pwscf.xml
2. 计算声子和电声相互作用
2.1 计算声子频率和变形势(deformation potential)
输入文件:ph.in
程序:ph.x (QuantumESPRESSO)
ph.in
Phonon
&INPUTPH
lshift_q = .true.
ldisp = .true.
search_sym = .false.
tr2_ph = 1.0d-15
alpha_mix = 0.3
fildvscf = 'dv'
nq1 = 4
nq2 = 4
nq3 = 3
nk1 = 8
nk2 = 8
nk3 = 6
/
$ mpiexec -np 29 PATH/ph.x -nk 29 -in ph.in > ph.out
重要的变量:
fildvscf = ‘dv’
变形势的文件名,下一步会用到
ldisp = .true.
在均匀q网格上计算声子。
lshift_q = .true.
移动q网格以避免Γ点的奇点。SCTK软件要求用四面体方法计算声子,这个是必须加的。
nq1, nq2, nq3
The q grid.
生成文件:
ph.out
pwscf.wfc* #一共144个,是节点所有的任务数ntask
matdyn0 #所有不可约q点的位置
matdyn* #共nq个,是每个q点的矩阵
_ph0/ #文件夹
_ph0/pwscf.phsave/#文件夹
_ph0/pwscf.phsave/control_ph.xml
_ph0/pwscf.phsave/dynmat.a.b.xml
#a是q点数,b是ph.out文件中所有声子mode的格式
例如对MgB2,a=12,b=9,另外b的值对于每个声子可能是不同的!
‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘’‘‘’‘’’‘’‘’’‘’
There are 9 irreducible representations
Representation 1 1 modes - To be done
Representation 2 1 modes - To be done
Representation 3 1 modes - To be done
Representation 4 1 modes - To be done
Representation 5 1 modes - To be done
Representation 6 1 modes - To be done
Representation 7 1 modes - To be done
Representation 8 1 modes - To be done
Representation 9 1 modes - To be done
、、、、、、、、、、、、、、、、、、、、、、、、、、、、、、、、、
_ph0/pwscf.phsave/patterns.*.xml #共nq个
_ph0/pwscf.phsave/status_run.xml
_ph0/pwscf.q_* #文件夹,共nq个
_ph0/pwscf.q_*/pwscf.dv* #不知道是什么,指标也很奇怪1,121,25,49,73,97
_ph0/pwscf.q_*/pwscf.wfc* #一共ntask个
_ph0/pwscf.q_*/pwscf.dv*/pwscf.save #文件夹
_ph0/pwscf.q_*/pwscf.dv*/pwscf.save/charge-density.dat
_ph0/pwscf.q_*/pwscf.dv*/pwscf.save/data-file-schema.xml
_ph0/pwscf.save #文件夹
_ph0/pwscf.save/charge-density.dat
2.2 计算电声相互作用
输入文件: epmat.in
Electron-phonon matrix
&INPUTPH
lshift_q = .true.
ldisp = .true.
search_sym = .false.
nq1 = 4
nq2 = 4
nq3 = 3
fildvscf = 'dv'
electron_phonon = 'scdft_input'
nk1 = 4
nk2 = 4
nk3 = 3
elph_nbnd_min = 3
elph_nbnd_max = 5
/
程序: ph.x (QuantumESPRESSO)
$ mpiexec -np 8 PATH/ph.x -nk 8 -in epmat.in > epmat.out
重要参数
electron_phonon = “scdft_input”
用变形势和动态矩阵( the dynamical matrix )计算电声顶点( the electron-phonon verte)。
TIPS:这里与QE正常求解电声相互作用不同。electron_phonon= ‘interpolated’
elph_nbnd_min, elph_nbnd_max
由于电子态( the electronic states )之间的电声相互作用只在费米面附近(in the vicinity of the Fermi surface)影响间隙方程(the gap equation),我们可以减少bands以减少计算成本( the numerical cost)。
bands 的最大值和最小值(中间必须包括费米能)可以用QE中的fermi_velocity.x得到。
补:得到bands的最大值和最小值
复制scf.in,scf.out,pwscf.save文件夹及赝势文件到自己的小机器
mpiexec -np 29 ~/software/sctk/bin/fermi_velocity.x -nk 29 -in scf.in > FS_vf.out
生成文件:
epmat.out
elph*.dat #共nq个
会读取或更改
_ph0/pwscf.phsave/patterns.*.xml
_ph0/pwscf.phsave/status_run.xml
_ph0/pwscf.q_*/pwscf.save/data-file-schema.xml
_ph0/pwscf.save/charge-density.dat
3. 计算屏蔽库仑相互作用(screened Coulomb interaction)/自旋涨落(Spin-fluctuation)
3.1 在稠密K网格下进行Non-SCF计算(Non-SCF calculation with a dense k grid)
输入文件: nscf.in
&CONTROL
calculation = 'nscf'
prefix='h3s',
pseudo_dir = '.'
outdir='./tmp'
/
&SYSTEM
ibrav = 0 ,
celldm(1) = 1.88972688,
nat = 4 ,
ntyp = 2 ,
ecutwfc = 60 ,ecutrho = 600,
occupations = 'tetrahedra_opt',
la2F = .true.,
nbnd = 15
/
&ELECTRONS
/
ATOMIC_SPECIES
H 1.00784 h_pbe_v1.4.uspp.F.UPF
S 32.0590 s_pbe_v1.4.uspp.F.UPF
CELL_PARAMETERS
-1.5622620063709813 1.5622620063709813 1.5622620063709816
1.5622620063709818 -1.5622620063709809 1.5622620063709813
1.5622620063709813 1.5622620063709816 -1.5622620063709811
ATOMIC_POSITIONS (crystal)
H 0.0000000000 0.5000000000 0.5000000000
H 0.5000000000 -0.0000000000 0.5000000000
H 0.5000000000 0.5000000000 0.0000000000
S 0.0000000000 0.0000000000 -0.0000000000
K_POINTS {automatic}
32 32 32 0 0 0
程序: pw.x (QuantumESPRESSO)
$ mpiexec -np 32 PATH/pw.x -nk 32 -in nscf.in > nscf.out
重要参数
必须保证这里的np大于第二步的np
calculation = “nscf”
执行non self-consistent calculation.
la2f = .true.
产生pwscf.a2F文件,保存 Kohn-Sham energy.
nbnd
对于计算极化函数(the polarization function),我们不得不去计算一些空态。但是当我们计算绝缘体的时候我们不需要计算太多空态。通常,空状态的数量与占据态(occupied states)的数量相同。
生成文件:
nscf.out
pwscf.a2Fsave
会重写pwscf.xml
会重写pwscf.wfc* 一共ntask个
所以如果这一步的ntask少于第二步用的ntask的话会只覆盖前面的,会出问题
会更新pwscf.save的内容
更新data-file-schema.xml
不更新charge-density.dat
更新pwscf.save/wfc*.dat #MgB2变为210个
#nscf.out中的
#number of k points= 210 (tetrahedron method)
#因为nscf.in用了密网格
3.2 计算屏蔽库仑相互作用下的波函数(Calculation of wave functions for the screened Coulomb interaction)
Input file: twin.in
Program: pw.x (QuantumESPRESSO)
$ bash PATH/twingrid.x 4 4 4 >> twin.in
$ mpiexec -np 32 PATH/pw.x -nk 32 -in twin.in > twin.out
(此处用粗网格)
重要参数
calculation = “bands”
产生k点使用twingrid.x,将其重定向到输入文件,如上所示。k点网格必须和 ph.x计算电声相互作用时input 使用的 nq1, nq2, and nq3 一样。然后运行pw.x
生成文件:
twin.out
更新pwscf.xml
更新pwscf.save/data-file-schema.xml
更新pwscf.save/wfc*.dat 的前96个
#96是粗网格里的k点个数,在twin.out中
#number of k points= 96
3.3 计算屏蔽库仑相互作用
Input file: sctk.in
Program: sctk.x
$ mpiexec -np 32 PATH/sctk.x -nk 32 -in sctk.in > kel.out
重要参数:
calculation = “kel”
计算 screened Couplmb / spin-fluctuation interaction.
nq1, nq2, nq3
They must be the same as the k in the previous step.
生成文件:
kel.out
vel*.dat #共nq个
4. SCDFT SCF calculation
算完声子(第二步)和屏蔽库伦相互作用(第三步)我们就可以算SCDFT了
Input file: sctk.in (Should be modified)
Program: sctk.x
$ export OMP_NUM_THREADS=32
$ mpiexec -np 1 PATH/sctk.x < sctk.in > tc.out
重要参数
calculation = “scdft_tc”
通过改变这部分,我们改变了计算的类型。这里,我们用二分法得到Tc(the bisection method)。
5.更多分析
5.1画费米面相关的图
文献的图:
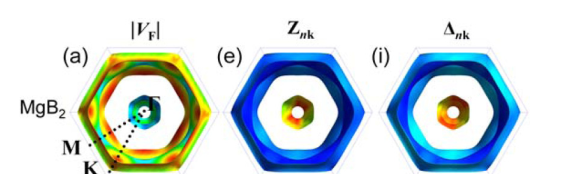
Top views of Fermi velocity of the electronic states |𝑉_𝐹 |(左),electron-phonon coupling renormalization 𝑍_𝑛𝑘(中), and superconducting gap function Δ_𝑛𝑘 (右)
现在我们来看怎么计算这三个图:
第一个图用QE/PP部分的fermi_velocity.x
http://www.quantum-espresso.org/Doc/pp_user_guide/node9.html
$ mpiexec -np 29 PATH/pw.x -nk 29 -in scf.in > F_scf.out #先重新执行一下自洽
$PATH/fermi_velocity.x scf.in > FS_vf.out
执行完会得到vfermi.frmsf文件,拖到桌面上,使用软件fermisurfer绘制图形

画第二个,第三个图:
Input file: sctk.in (Should be modified)
Program: sctk.x
$ mpiexec -np 1 PATH/sctk.x < sctk.in
calculation = “deltaf”
执行完会得到Z.frmsf,delta.frmsf文件,拖到桌面上,使用软件fermisurfer绘制图形

5.2其他分析
calculation = “lambda_mu_k”
生成 lambda.frmsf, mu.frmsf,使用软件fermisurfer可以绘制图形
输入文件不需要&KEL,只需要&SCDFT
calculation = “scdft”
在某个温度下执行SCDFT计算,然后输出一个文件delta.dat,该文件由以下后处理使用。
temp
温度(开尔文)。如果它被设为0或负值,sctk.x使用特殊的算法来精确的零开尔文
calculation = “qpdos”
准粒子DOS。它需要很长的计算时间。
5.3所有输入文件的k,q设置
https://arxiv.org/pdf/1911.06481.pdf
Sec.3 D 使用三种不同的网格密度
1.粗网格(C)
2.中等密度网格(M)
3.密网格(D:w)
下面是例子中的输入文件使用的网格
| 文件名 | Al | MgB2 |
|---|---|---|
| 1.scf.in | k=888(M);q=无 | k=886;q=无 |
| 2.1 ph.in | k=888(M);q=444( C) | k=886;q=443 |
| 2.2 epmat.in | k=444( C);q=444( C) | k=443;q=443 |
| 3.1 nsfc.in | k=16 16 16(D);q=无 | k=16 16 12 q=无 |
| 3.2 twin.in | k=444(C) | k=443 |
| 3.3 sctk.in | q=444( C) | q=443 |
5.4 画声子谱
和正常画声子谱一样,注意一定要用sctk里的q2r.x和matdyn.x直接用QE里的会报错。且q2r.in和matdyn.in里要加一行lshift_q = .true.
6. sctk.in的参数设置
6.1 calculation = “kel”


start_q : 计算屏蔽库伦相互作用的第一个q点
last_q:最后一个q点,默认是不可约BZ中的q点数
laddxc:使用的假设 0 : RPA, 1: Adiabatic LDA.
lsf:是否计算the spin-fluctuation,1计算,0不计算
ecutwfc:和pw.x一样
nq1, nq2, nq3:和 twingrid.x 一样
nci:The number of Matsubara frequencies of screened Coulomb interactions.
在程序中对于它的描述为“# of Chebyshev int. : nci”(切比雪夫整数???)
6.2 calculation = “scdft”

temp: 温度(开尔文)
fbee:计算gap函数的第一个带
lbee:计算gap函数的最后一个带。nbnd in pw.x
xic:间隙函数外推法的参数。小于0时不使用这个方法。默认不使用。
nmf:计算 the Coulomb kernel的松原频率数。设为0时使用静态Coulomb
kernel。设成负数只用the phononic kernel,Coulomb kernel变为0.
nx:费米能级附近辅助能量网的数目。
ne:仅用于准粒子DOS( calculation of quasi-particle DOS.网站上关于它有专门的章节)的计算。准粒子态密度的能量网格数。
emin:辅助能源网最小能量尺度参数。单位:Ry
emax:仅用于准粒子DOS的计算。计算QPDOS的最大能量。单位:meV
electron_maxstep:求解gap方程的最大迭代次数
conv_thr:gap方程的收敛阈值,当新间隙函数和旧间隙函数之差的范数小于它时,认为系统是收敛的.单位:Ry
filedyn:It must be the same as filedyn in ph.x.别写最好。是动力学矩阵存储的名称。
spin_fluc:是否包含自旋涨落
6.3 是否包含SF的设置
这是MgB2例子的结果
| Tc | mu | lambda | |
|---|---|---|---|
| kel,scdft,lambda三个步骤全部包含SF | 12.308 | 0.29893 | 0.58202 |
| kel包含,scdft包含,lambda不包含 | 12.308 | 0.23894 | 0.58202 |
| kel包含,scdft不包含,lambda不包含 | 16.686 | 0.23894 | 0.58202 |
| kel,scdft,lambda全不包含 | 16.686 | 0.23894 | 0.58202 |
kel这一步是否包含没有影响,scdft不包含会影响超导温度,lambda不包含会影响mu,这些参数都不会影响lambda
















 1633
1633











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









