







Github链接:GitHub - AntonBankevich/LJA
操作环境:CentOS 8.5 64位
零、引言介绍
LJA是最近发表在nature子刊biotechnology的一个三代测序组装软件,集合并应用了the Bloom filter, the rolling hash, the sparse de Bruijn graph and the disjointig generation等四种算法。它首先利用disjointig generation算法构建 稀疏de Bruijn graph,之后通过更改k值的方法纠正读取错误
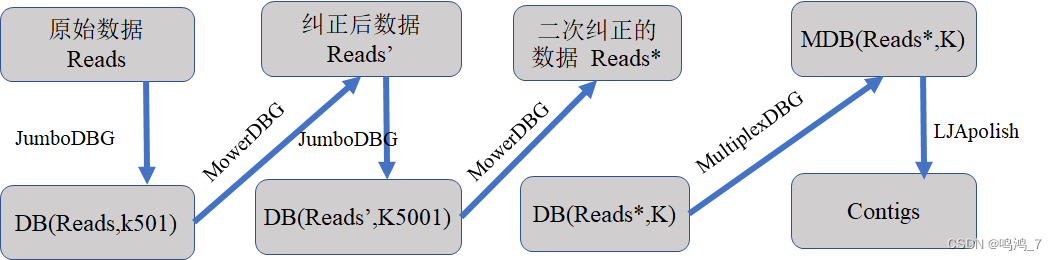
LJA流程图
一、安装环境
首先是作者要求的环境,要求cmake(3.1及以上),gcc(7.0及以上,要支持c++17的编译运行),zlib,以及GNU的版本要很高(要有支持c及c++的运行环镜)
1、安装cmake(3.24)
cmake提供编译好的包,建议下载下来放到环境里就可以了
#将cmake加载到环境里
wget https://cmake.org/files/v3.24/cmake-3.24.0-linux-x86_64.tar.gz#这里安装的是cmake3.24版本
tar xvf cmake-3.24.0-linux-x86_64.tar.gz #解压
vim ~/.bashrc
export PATH=your/cmake-3.24.0-linux-x86_64/bin:$PATH
vim ~/.bashrc #将cmake加载到环境变量里
cmake --version #查看cmake版本,
#成功将cmake加载到环境里2、安装gcc(7.0.4)
首先要检查系统是否支持gcc以及g++环境
gcc --version#检查gcc版本
g++ --version#检查g++版本如果都支持,且两者版本都高于7,可以跳转到第3步
一般而言系统都会自带gcc版本,不过不会太高(4.8左右),而LJA需要gcc版本至少为7,所以需要去安装高版本gcc
接下来安装gcc9.1.0版本
//由于源码编译时间非常长(8小时及以上)且十分容易报错,建议先去下载编译好的gcc的包,如果用不了再使用源码安装
wget http://ftp.gnu.org/gnu/gcc/gcc-9.1.0/gcc-9.1.0.tar.gz
tar -xvf gcc-9.1.0.tar.gz
cd gcc-9.1.0
./contrib/download_prerequisites
# 等待安装依赖成功
mkdir gcc-9.1.0 # 编译路径
cd gcc-9.1.0
../configure --disable-checking --enable-languages=c,c++ --disable-multilib --prefix=your_path --enable-threads=posix --with-system-zlib
make # 时间非常长
make install
#安装成功后
gcc --version
g++ --version #成功3、安装zlib
wget http://www.zlib.net/fossils/zlib-1.2.11.tar.gz
tar -zxvf zlib-1.2.11.tar.gz
cd zlib-1.2.11
mkdir build #建议分开安装,避免出现问题
./configure --perfix=/dellfsqd2/ST_OCEAN/USER/xuqi/software/zlib-1.2.11/build #如果没有root权限,则安装到指定位置
make & make install4、下载LJA源码,编译安装LJA
按照作者提供的方式安装即可

#把bin添加到环境,之后就可以直接使用lja了
export PATH=/dellfsqd2/ST_OCEAN/USER/xuqi/software/LJA/bin:$PATH
二、LJA软件运行及比对分析
1、下载数据
以SRR11606869数据为例(作者文章中使用的),这是玉米的PacBio HiFi测序数据
直接用服务器下载(也可以用迅雷,然后用xftp传到服务器上)(时间极长,网速慢,大概12个小时)
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR11606869/SRR116068692、数据格式转换
lja支持输入为fasta和fastq格式的序列,但我们从sra上下载的数据格式是sra,因此需要进行转换,具体流程看这里
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz
tar xzvf sratoolkit.current-centos_linux64.tar.gz
cd sratoolkit.2.9.2-centos_linux64
export PATH=/dellfsqd2/ST_OCEAN/USER/xuqi/software/sratoolkit.2.9.2-centos_linux64/bin:$PATH #依旧是放到环境下
#使用的工具是fastq-dump.2.9.2
#直接使用就好了
fastq-dump.2.9.2 SRR116068693、数据FastQC质检
尽管是NCBI的数据,但对数据质量评估是必不可少的
nohup fastqc SRR11606869.fastq -t 10 -o fastqc/ &
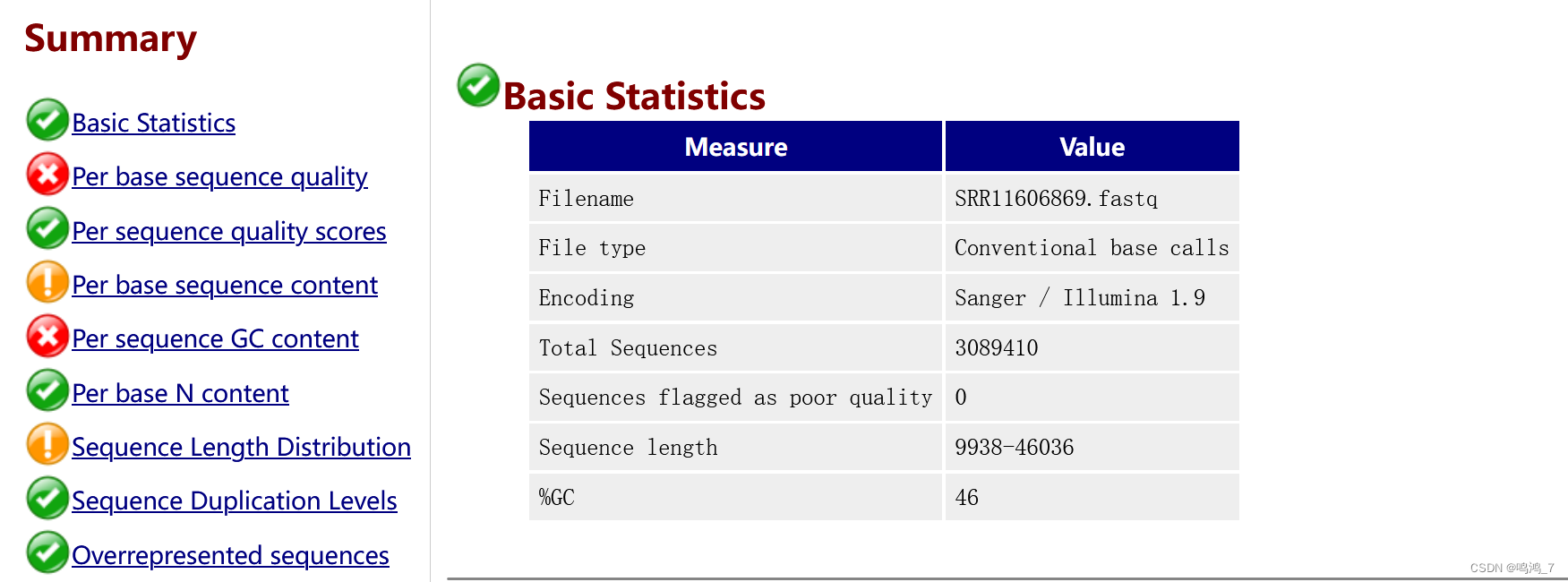
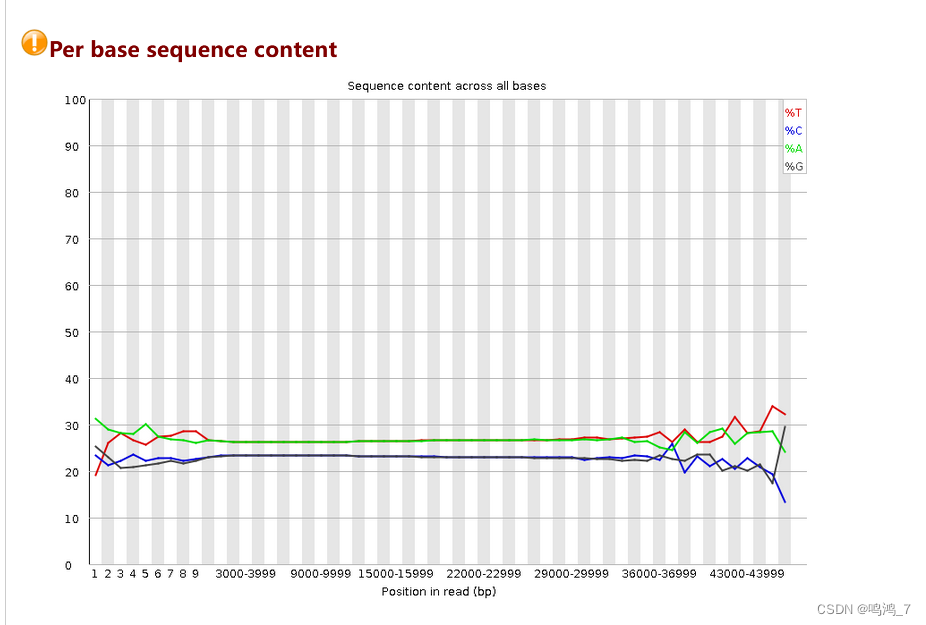
主要是起始端和末端的碱基有问题,应该去掉(Trimmomatic数据过滤),补充代码如下,有时间再来一遍
java -jar trimmomatic-0.38.jar SE -phred64 -trimlog test.log.txt test.fastq
test.processed.fastq ILLUMINACLIP:adapters/TruSeq3-SE.fa:2:30:10 LEADING:3
TRAILING:3 MINLEN:25
#最原始代码,需要修改参数4、运行lja
尽管lja可以调整参数,但作者建议使用默认参数运行。lja单独使用即可得到最终组装结果(lja有三个模块,只在运行的时候才会体现出来;第一个模块jumboDBG可以单独使用,会输出图以及disjointigs序列)
所以就直接运行就好了,默认是16线程(一定要加 nohup!!!!!!! )。玉米是二倍体物种,加上参数--diploid
nohup lja -o new/ --diploid --reads SRR11606869.fastq &
过程大概要40个小时及以上
#可以时不时的查看dbg.log,掌握进度
cat dbg.log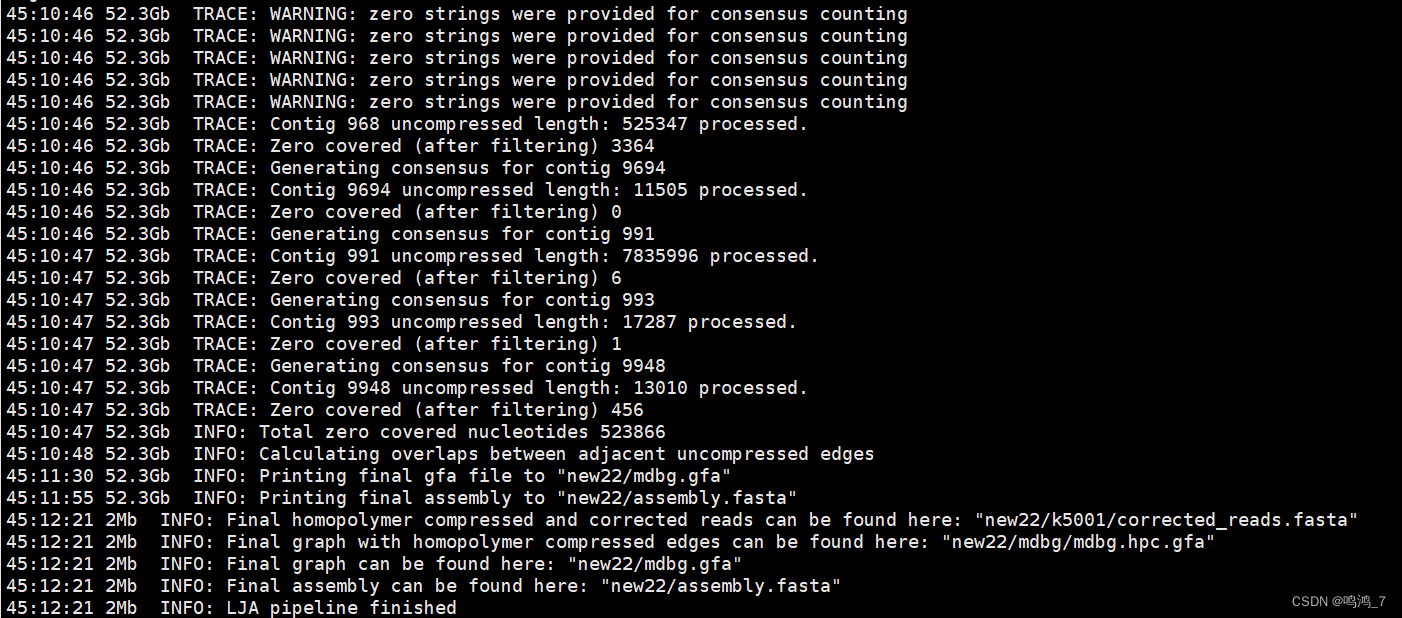
最后可以得到assembly.fasta,也就是组装好的序列
以及lja生成的文件目录

5、QUAST评估lja基因组组装质量
教程可以看 这一篇 ,也是很常规的安装
wget -c https://github.com/ablab/quast/releases/download/quast_5.1.0rc1/quast-5.1.0rc1.tar.gz
tar -zxvf quast-5.1.0rc1.tar.gz
python quast.py --help
python quast.py --version
# QUAST v5.1.0rc1, 6260eff0
首先下载好玉米的参考基因组以及gff格式文档(推荐迅雷)
#玉米参考基因组
wget http://ftp.ensemblgenomes.org/pub/plants/release-7/fasta/zea_mays/dna/Zea_mays.AGPv2.dna.toplevel.fa
#玉米参考gff3
wget http://ftp.ensemblgenomes.org/pub/plants/release-7/gtf/zea_mays/zea_mays.AGPv2.60.gtf然后就可以进行比对了(时间也比较长,不过还好,几个小时而已)
python3.4 geno/quast-5.1.0rc1/quast.py new22/assembly.fasta -r geno/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.dna.toplevel.fa.gz -g geno/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.54.gff3.gz -o result2 -t 26、hifiasm组装,做对比实验
#运行hifiasm
nohup /dellfsqd2/ST_OCEAN/USER/xuqi/software/hifiasm/hifiasm -o kk -t 15 SRR11606869.fastq &
7、QUAST评估hifiasm基因组组装质量
首先要将gfa格式转为fasta格式,hifiasm官网给了解决方案
awk '/^S/{print ">"$2;print $3}' hifiasm.bp.p_ctg.gfa > hifiasm.p_ctg.fasta 然后比对
nohup python3.4 geno/quast-5.1.0rc1/quast.py hifiasm.p_ctg.fasta -r geno/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.dna.toplevel.fa -g geno/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.54.gff3.gz -o resultx -t 2 &等很久就可以了
三、结果展示
对玉米SRR11606869的组装评估
| 指标 | LJA结果 | hifiasm结果 |
| Total length (>= 0 bp) | 2136MB | 2170MB |
| # contigs | 1064 | 731 |
| # contigs (>= 50K bp) | 338 | 440 |
| # misassemblies | 470 | 1476 |
| # misassembled contigs | 124 | 335 |
| Genome fraction (%) | 97.533 | 98.278 |
| Duplication ratio | 1.004 | 1.012 |
| Largest alignment | 59MB | 89MB |
| Total aligned length | 2133MB | 2165MB |
| N50 | 22MB | 45MB |
| L50 | 24 | 16 |
| NA90 | 3.3MB | 3.1MB |
| Unaligned length | 0.31MB | 0.33MB |
各有优劣,不过hifiasm更好一点,猜测可能是lja现在还不完善以及没有准确的参考基因组。LJA的错误率更低,原因是经历了两轮纠错


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









