





摘要
本研究提出了一种基于深度学习模型的方法ncDENSE,通过提取ncRNAs序列特征来预测ncRNAs家族。对ncRNAs序列中的碱基进行one-hot编码,并将其输入到包含动态双向门控循环单元( Bi-GRU )、密集卷积网络( DenseNet )和注意力机制( AM )的集成深度学习模型中。具体来说,动态Bi-GRU用于提取上下文特征信息,捕获ncRNAs序列的长期依赖关系。利用AM对Bi-GRU提取的特征赋予不同的权重,将注意力集中在权重较大的信息上。而DenseNet则用于提取ncRNAs序列的局部特征信息,并通过全连接层进行分类。
本文对比了四种具有代表性的深度学习模型,包括深度神经网络( DNN )、卷积神经网络( CNN )、循环神经网络( RNN )以及循环和卷积神经网络的组合( RNN + CNN )。图2数据表明,对于不同长度的ncRNAs序列,RNN + CNN模型普遍优于其他3种模型,因此选择RNN + CNN模型进行ncRNAs家族预测。该模型包括动态BiGRU、注意力机制和DenseNet,与现有方法相比,不仅简化了预测过程,而且提高了准确率。
方法
数据采集与处理
本文处理的数据均来自Rfam数据库,该数据库包含13个ncRNAs家族(即microRNAs , 5S _ rRNA , 5.8S _ rRNA ,核酶, CD-box , HACA-box , scaRNA , tRNA , Intron _ gpI , Intron _ gpII , IRES , leader和核糖开关),共6320条非冗余ncRNAs数据,其中IRES数据320条,其余ncRNAs家族各500条。 ncDENSE在模型训练过程中采用十折交叉验证法,将每个家族的ncRNAs数据分为10份,其中9份作为训练集,剩余1份作为测试集。最后将10个交叉验证法的结果取平均值。为了将ncRNAs序列输入到集成深度学习模型中,本文使用one - hot编码将每个ncRNA的碱基编码为1 * 4、1 * 8和1 * 12数据[ 31 ]。A (腺嘌呤)、U (尿嘧啶)、G (鸟嘌呤)和C (胞嘧啶)代表四种常见ncRNA的碱基,其编码规则如表2所示,' N '代表一些稀有碱基。
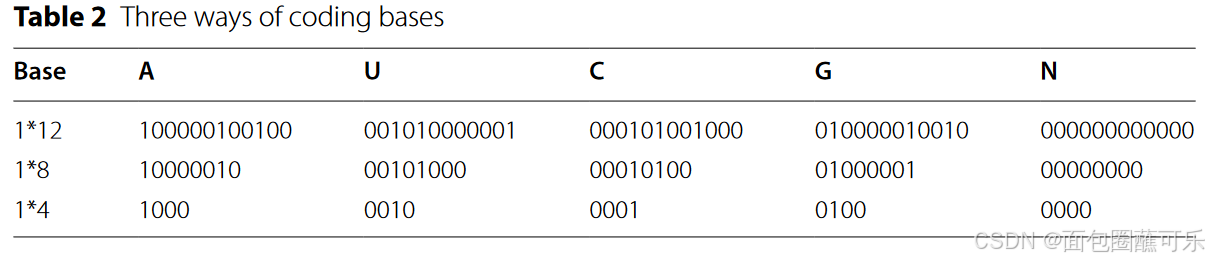
根据比较,1 * 8编码方法比1 * 4和1 * 12编码方法更有效,因此在ncDENSE中使用1 * 8独热编码方法对ncRNAs序列的碱基进行数字编码,编码后每个ncRNAs序列的长度为L * 8 ( L是ncRNAs序列中的碱基数)。
方法
ncDENSE方法使用了由动态Bi - GRU、Attention Mechanism和DenseNet三个组件组成的集成深度学习模型。动态Bi - GRU主要负责捕获ncRNAs序列的长期依赖关系,以提取其上下文特征信息。同时,Attention机制主要作用是为Bi - GRU提取的特征根据其重要性分配不同的权重。而DenseNet则通过密集连接的方式提取ncRNAs序列的局部特征。最后,将动态Bi - GRU、Attention Mechanism和DenseNet提取的ncRNAs序列特征输入到全连接层进行ncRNAs家族分类。本文提出的ncDENSE方法将每个批次的ncRNAs序列数量设置为32个。ncDENSE方法的主要工作原理如图12所示。
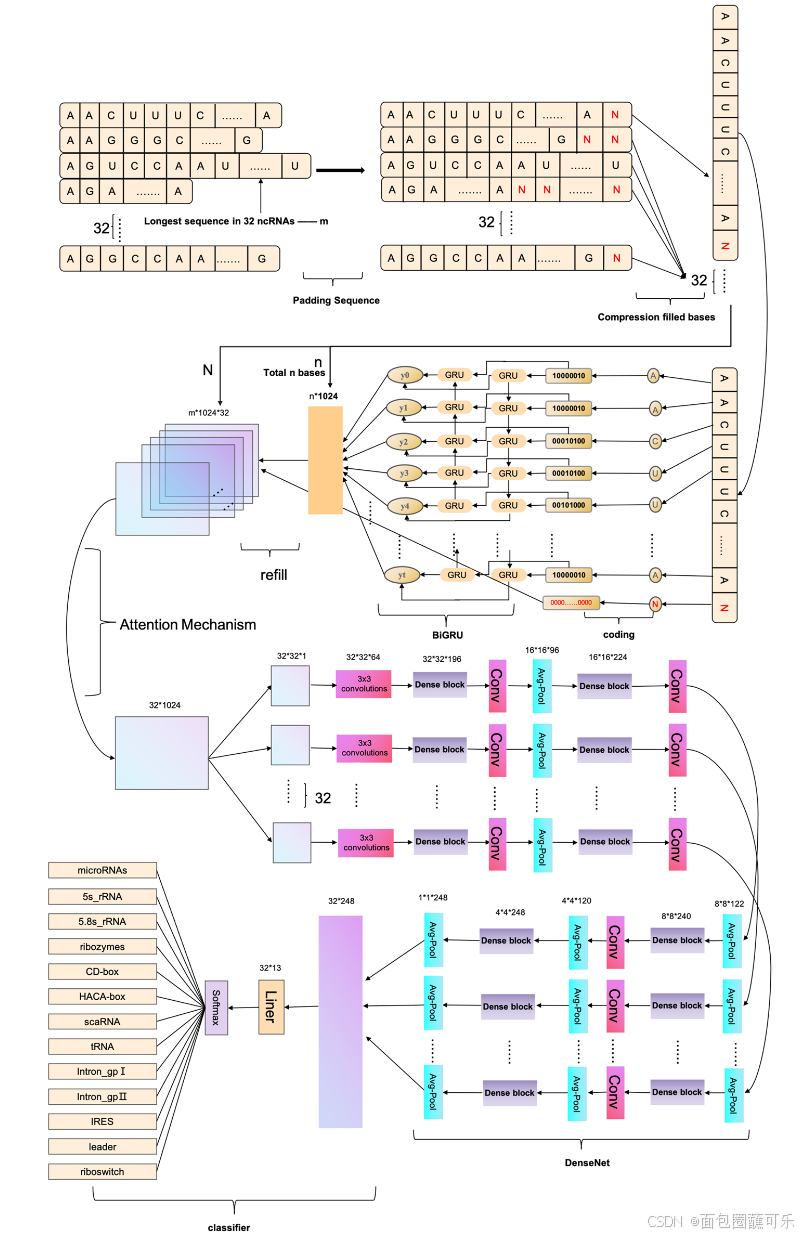
( 1 )动态Bi - GRU
动态Bi - GRU摒弃了传统的ncRNAs序列的填充和切割,将不同长度的ncRNAs序列中的所有碱基信息输入到Bi - GRU中,避免了ncRNAs序列中碱基信息的丢失或无用信息的增加。因此,防止了Bi - GRU模型提取ncRNAs序列的冗余特征信息或丢失ncRNAs序列的重要特征信息。ncRNAs序列是具有紧密关联的上下文信息的数据。双向RNN模型通过门控信息可以很好地应用于提取ncRNAs序列的上下文特征,不仅可以保留每个碱基之前的碱基信息,还可以记录当前碱基之后的碱基信息。动态Bi - GRU的工作原理如图12所示。GRU是LSTM的变体,比LSTM的结构更简单,可以有效解决RNN网络中的长依赖问题。GRU单元只有两个门信息,包括更新门和复位门,可以提高模型的计算效率。GRU单元结构示意图如图13所示,其工作原理可分为以下4个部分。
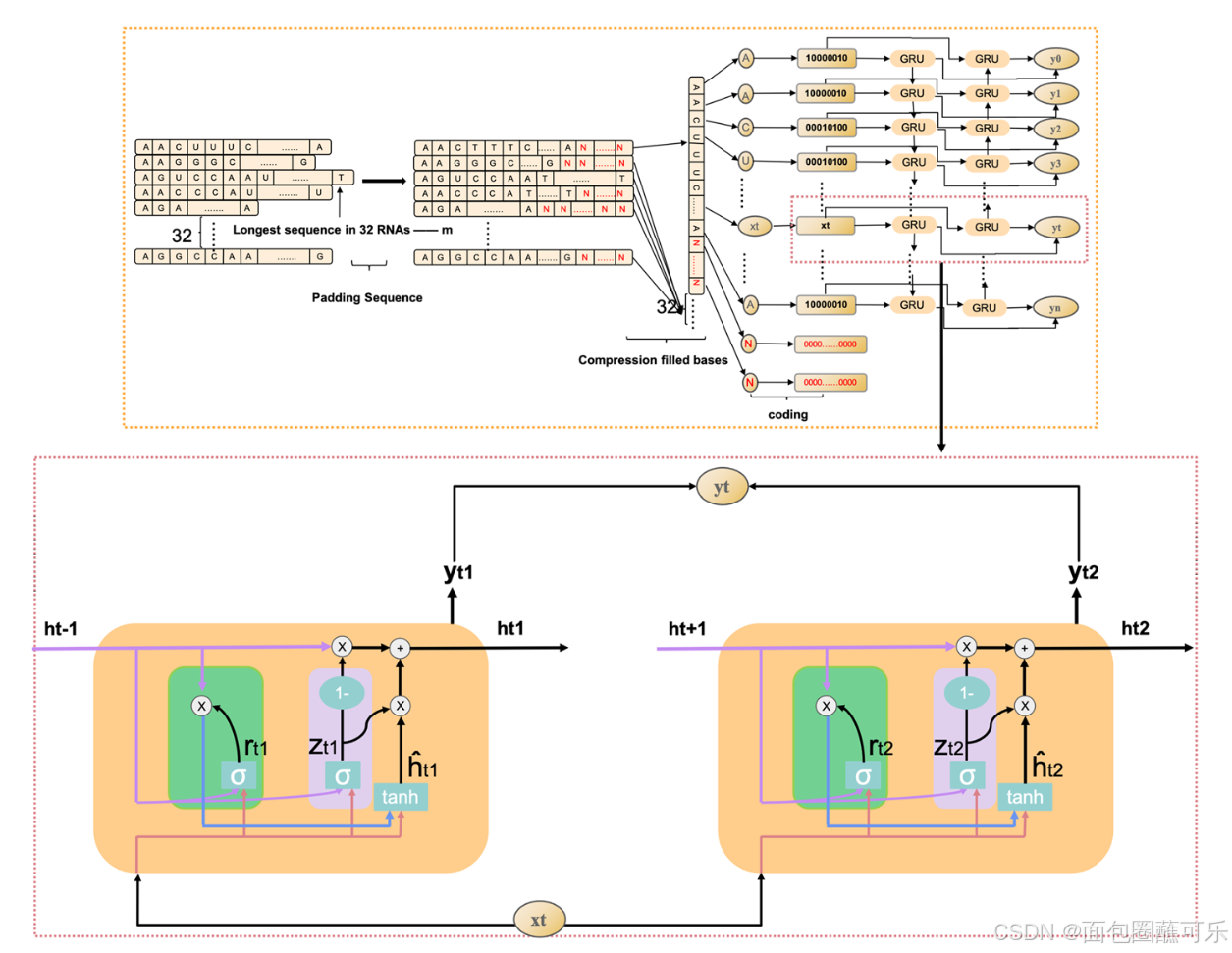
序列长度为32的RNA-填充其他序列-压缩填充序列-碱基编码-BiGRU提取序列的上下文信息
( 1 )复位门(图13中的绿色部分代表复位门)的计算复位门决定了新的输入是如何与之前的信息相结合的。更大的复位门值代表需要从前一时刻记住更多的信息,更多的新输入( Xt )与前一时刻的记忆( ht-1 )相结合。相反,一个较小的复位门值表明从前一时刻应该记住较少的信息,较少的新输入( Xt )应该与前一时刻的记忆( ht-1 )相结合。其余门值可由公式计算得到公式6。
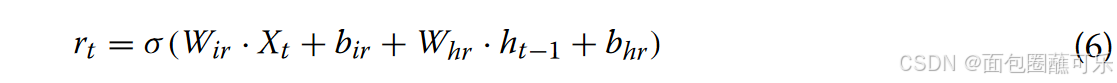
其中Wir和Whr表示复位门的权重矩阵,bir和bhr表示复位门的偏差,Xt表示t时刻的信息输入,ht-1表示t-1到t时刻的信息传递,σ表示激活函数sigmoid。
( 2 )更新门(图13中的粉红色部分代表了更新门)的计算更新门用来控制上一时刻的状态信息被带入当前状态的程度。更新门数值越接近于1,表明新的输入留下了更多的数据,越接近于0,表明新的输入被遗忘的越多。更新门值可由公式计算得到公式7。
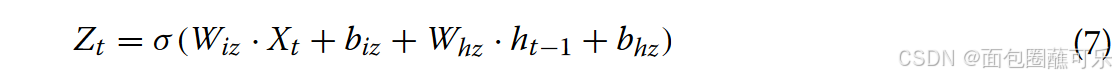
其中Wiz和Whz是更新门的权重矩阵,biz和bhz分别是更新门的偏差。
( 3 )当前记忆内容的重置
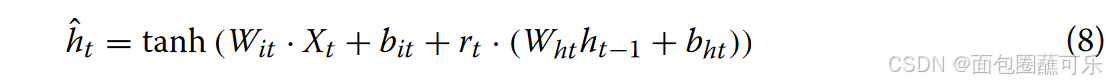
其中Wit和Wht表示权重矩阵,bit和bht表示偏差,表示当前时刻计算后的信息。
( 4 ) GRU输出计算

其中表示从上一时刻传递下来的信息在
中遗忘,并在当前时刻的
中增加一些信息,形成传递到下一时刻的最终记忆。
( 2 )注意力机制
注意力机制的核心思想是为重要的输入信息分配更多的权重,从而使注意力可以巧妙而合理地转移到忽略无关信息和放大重要信息上。这样,在注意力集中的区域,信息接收的灵敏度和处理速度都得到了极大的提高。注意力机制的工作原理如式( 10 )所示。

其中C是注意力机制的输出,Lx表示输入RNA序列的长度,a表示分配给第j个隐藏状态的系数,h表示第j个隐藏状态。注意力机制的特点是能够很好的完成将ncDENSE聚焦在ncRNA家族重要片段上的任务。
( 3 ) DenseNet
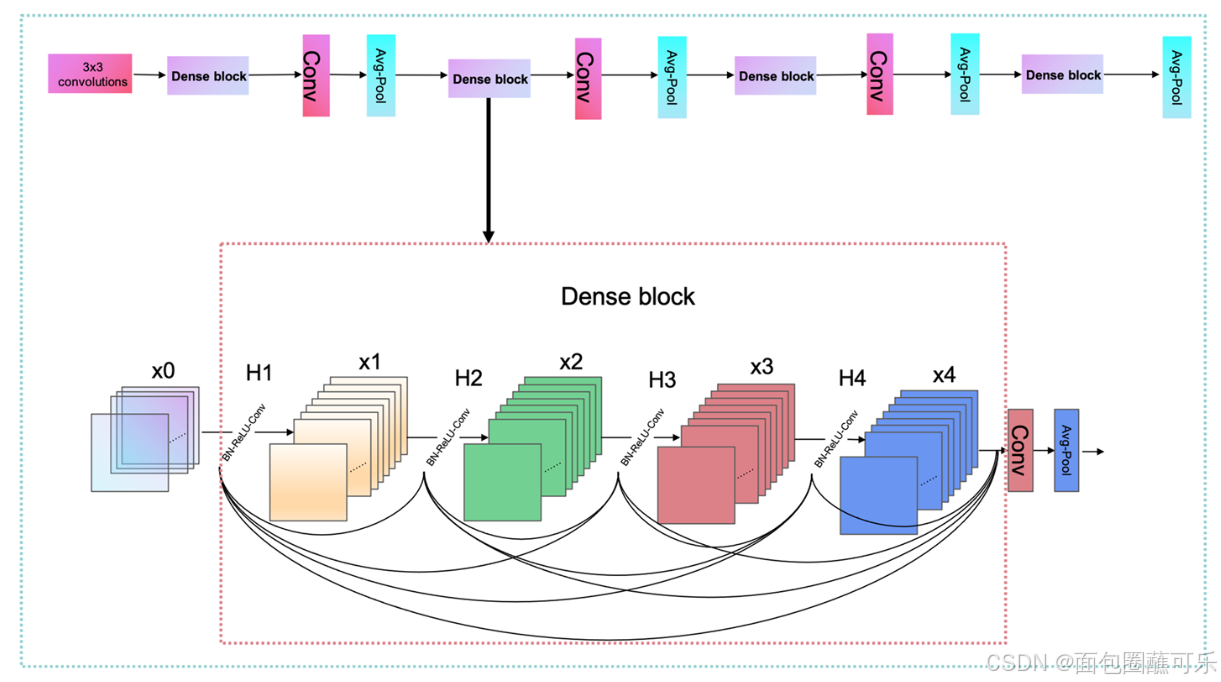
DenseNet的设计受到ResNet的启发。通过改进ResNet的连接方式,在DenseNet中提出了一种密集连接机制。密集连接机制的主要思想是将所有层相互连接,每一层都接收来自之前所有层的信息作为输入。本文采用4层Dense块,每个Dense块共有10个连接,如图14所示。各层的计算如式(11)所示。
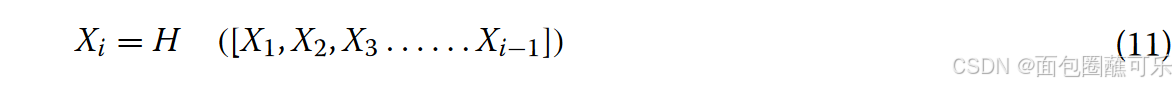
在公式(11)中Xi是第i层的结果,H采用方程中BN + ReLU + 3 * 3CONV的结构。每个ncRNA序列由4个密集块提取,4个密集块输出特征的大小分别为32 * 32 * 196、16 * 16 * 224、8 * 8 * 240、4 * 4 * 248。过渡层由Conv层和Avg Pool层组成,可以对密集块输出的特征进行降尺度,降尺度后3个过渡的维度分别为16 * 16 * 96、8 * 8 * 122、4 * 4 * 120。Avg-Pool最后一层的大小为1 * 1 * 248,最后32个ncRNAs序列被PyTorch中的Liner整形为32 * 248用于全连接层的分类。
结果
ncRNAs的序列结构、平面结构和空间结构包含了ncRNAs家族的特征,可以作为RNN+CNN集成深度学习模型的输入。ncRNAs的一级结构具有家族一致的序列片段,因此可以利用深度学习模型提取ncRNAs序列特征,之后主要通过ncRFP和ncDLRES对未知ncRNAs所属家族进行分类。与ncRFP相比,本文提出的方法在编码后将ncRNAs的碱基序列动态地输入到动态Bi-GRU模型中,避免了对ncRNAs序列的剪切和填充,保留了ncRNAs序列的全部特征信息。相对于ncDLRES,ncDENSE使用Bi-GRU代替LSTM,LSTM具有3个门信息,而GRU只保留2个门信息,从而显著提高了代码的运行效率。Bi-GRU属于双向RNN模型,不仅保留了ncRNAs序列当前碱基之前的碱基信息,还记录了当前碱基之后的碱基信息,有助于更好地提取ncRNAs序列特征。在ncDLRES方法中,ResNet用于提取ncRNA序列的局部特征信息,本文使用的DenseNet模型较好地解决了深度网络反向传播时梯度消失的问题。与基于ncRNAs二级结构的ncRNAs家族预测方法相比,本文提出的方法直接跳过ncRNAs二级结构的预测,不仅提高了预测精度,而且提高了预测效率。
与之前提出的方法相比,ncDENSE方法在结果数据上有两个方面的显著提升。第一种改进体现在整体数据上,通过十折交叉验证的平均值比较各方法的数据;第二种改进体现在单个ncRNA家族上,通过比较不同方法预测的13个ncRNA家族的性能。
本文提出了一种新的融合动态BiGRU + Attention Mechanism + DenseNet模型的ncDENSE方法。一方面,通过不填充或切割ncRNAs序列的方式对动态Bi-GRU进行改进,使得ncRNAs的序列特征都可以输入到Bi-GRU模型中。另一方面,Bi-GRU是一个双向的RNN模型,它保留了当前碱基之前的碱基信息,并记录了当前碱基之后的碱基信息,从而可以充分提取ncRNAs序列的上下文特征信息。此外,注意力机制可以为ncRNAs序列的重要特征分配更多的权重,从而可以巧妙而合理地调整注意力以转移,从而忽略无关信息并放大重要信息。更进一步,DenseNet利用稠密连接性更好地解决了深度网络中反向传播梯度消失的问题。
结论
对ncRNAs家族的预测可以初步判断ncRNAs的功能。面对海量的高通量ncRNA序列数据,基于生物实验的方法无法满足预测的需求,而现有的主要计算方法存在过程复杂、精度不高等问题。本文提出了一种新的ncRNAs家族预测的计算方法ncDENSE。总的来说,ncDENSE有四个优点。
第一:ncDENSE方法通过提取ncRNAs的序列特征来预测ncRNAs家族,跳过了获取ncRNAs二级结构的过程,提高了ncRNAs家族预测精度,简化了预测过程。
第二,ncDENSE方法摒弃了传统的对ncRNAs序列的填充和分割,将ncRNAs序列的全部特征全部输入到深度学习模型中。
第三:ncDENSE方法采用双向RNN模型-Bi-GRU,与单向RNN模型相比,该模型可以保留当前碱基之前的碱基信息,并记录当前碱基信息之后的碱基信息,从而有利于ncRNAs序列上下文特征的提取。
第四:ncDENSE方法利用DenseNet网络提取ncRNAs序列的局部特征,有助于通过密集连接更好地缓解深度网络中反向传播的梯度消失问题。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









