






上一篇讲了Geatpy的快速入门:https://blog.csdn.net/qq_33353186/article/details/82014986
但是光是几个例子是远远不能熟练掌握python遗传和进化算法编程的,得进一步了解其原理以及API。
Geatpy提供面向对象的简单封装的开放式进化算法框架,可以方便、自由地与其他算法或项目相结合。Geatpy的文件结构如下图所示:
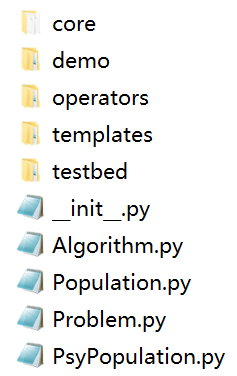
其中“core”文件夹里面全部为Geatpy工具箱的内核函数;“templates”文件夹存放的是Geatpy的进化算法模板;“testbed”是进化算法的测试平台,内含多种单目标优化、多目标优化、组合优化的测试集。“demo”文件夹中包含了应用Geatpy工具箱求解问题的案例。“operators”是2.2.2版本之后新增的,里面存放着面向对象的重组和变异算子类,这些重组和变异算子类通过调用“core”文件夹下的重组和变异算子来执行相关的操作。
Geatpy 的面向对象进化算法框架有四个大类:Algorithm(算法模板顶级父类)、Population(种群类)、PsyPopulation(多染色体种群类) 和Problem(问题类),分别存放在“Algorithm.py”、“Population.py”、“Problem.py”文件中。其UML 类图如下所示:
Geatpy 的面向对象进化算法框架有四个大类:Algorithm(算法模板顶级父类)、Population(种群类)、PsyPopulation(多染色体种群类) 和Problem(问题类),分别存放在“Algorithm.py”、“Population.py”、“Problem.py”文件中。其UML 类图如下所示:

Problem类定义了与问题相关的一些信息,如问题名称name、优化目标的维数M、决策变量的个数Dim、决策变量的范围ranges、决策变量的边界borders等。maxormins是一个记录着各个目标函数是最小化抑或是最大化的list类型列表,其中元素为1表示对应的目标是最小化目标;为-1表示对应的是最大化目标。例如M=3,maxormins=[1,-1,1],此时表示有三个优化目标,其中第一、第三个是最小化目标,第二个是最大化目标。varTypes是一个记录着决策变量类型的行向量,其中的元素为0表示对应的决策变量是连续型变量;为1表示对应的是离散型变量。待求解的目标函数定义在aimFunc()的函数中。calReferObjV()函数则用于计算或读取目标函数参考值(一般用理论上的目标函数的最优值作为参考值),该参考值可以用于后续的指标分析。在实际使用时,不是直接在Problem类的文件中修改相关代码来使用的,而是通过定义一个继承Problem的子类来完成对问题的定义的。这些在后面的章节中会详细讲述。getReferObjV()是Problem父类中已经实现了的一个函数,它先尝试读取特定文件夹中的目标函数值参考数据,如果读取不到,则调用calReferObjV()进行计算。对于Problem类中各属性的详细含义可查看Problem.py源码。
Population类是一个表示种群的类。一个种群包含很多个个体,而每个个体都有一条染色体(若要用多染色体,则使用PsyPopulation类)。除了染色体外,每个个体都有一个译码矩阵Field(或俗称区域描述器)来标识染色体应该如何解码得到表现型,同时也有其对应的目标函数值以及适应度。种群类就是一个把所有个体的这些数据统一存储起来的一个类。比如里面的Chrom是一个存储种群所有个体染色体的矩阵,它的每一行对应一个个体的染色体;ObjV是一个目标函数值矩阵,每一行对应一个个体的所有目标函数值,每一列对应一个目标。对于Population类中各属性的详细含义可查看Population.py源码以及下一章“Geatpy数据结构”。
PsyPopulation类是继承了Population的支持多染色体混合编码的种群类。一个种群包含很多个个体,而每个个体都有多条染色体。用Chroms列表存储所有的染色体矩阵(Chrom);Encodings列表存储各染色体对应的编码方式(Encoding);Fields列表存储各染色体对应的译码矩阵(Field)。
Algorithm类是进化算法的核心类。它既存储着跟进化算法相关的一些参数,同时也在其继承类中实现具体的进化算法。比如Geatpy中的moea\_NSGA3\_templet.py是实现了多目标优化NSGA-III算法的进化算法模板类,它是继承了Algorithm类的具体算法的模板类。关于Algorithm类中各属性的含义可以查看Algorithm.py源码。这些算法模板通过调用Geatpy工具箱提供的进化算法库函数实现对种群的进化操作,同时记录进化过程中的相关信息,其基本层次结构如下图:

Geatpy工具箱提供了大量的跟进化算法有关的内核函数,涵盖了单目标优化、多目标优化、组合优化等方面的众多算子,同时提供指标评价、绘图等功能性函数。下图展示了Geatpy的函数调用关系。中心是进化算法模板,它调用高级的运算函数(selecting, recombin, mutate)来实现进化优化。高级函数进一步调用相关的低级运算函数(即实现选择、交叉、变异等底层算法的函数)。这种层级调用关系使Geatpy的结构十分清晰,更重要的是,你可以自定义更多低级运算函数来轻松自由地扩展Geatpy。

这些库函数具体有:
1. 与初始化种群相关的函数
• crtfld (生成译码矩阵,俗称“区域描述器”)
• crtbp (创建二进制种群染色体矩阵)
• crtip (创建元素是整数的种群染色体矩阵)
• crtpp (创建排列编码种群染色体矩阵)
• crtrp (创建元素是实数的种群染色体矩阵)
• meshrng (网格化决策变量范围)
2. 进化迭代相关函数
当完成了种群染色体的初始化后,就可以进行进化迭代了。这部分是在进化算法模
板里调用。迭代过程中包括:
• 调用ranking 或scaling 等计算种群适应度。
• 调用selecting 进行选择操作(也可以直接调用低级选择函数)。
• 调用recombin 进行重组操作(也可以直接调用低级重组函数)。
• 调用mutate 进行重组操作(也可以直接调用低级变异函数)。
3. 适应度计算
• ranking (基于等级划分的适应度分配计算)
• scaling (线性尺度变换适应度计算)
• indexing (指数尺度变换适应度计算)
• powing (幂尺度变换适应度计算)
对于多目标进化优化,由于各种多目标优化算法所采用的适应度计算方法门类有很多,因此此时的适应度计算交由继承了Algorithm 的具体算法模板类中实现,详见相关源码。
4. 选择
selecting 是高级选择函数,它调用下面的低级选择函数:
• dup (Duplication,基于适应度排序的直接复制选择)
• ecs (Elite Copy Selection,精英复制选择)
• etour (精英保留锦标赛选择)
• otos (One-to-One Survivor Selection,一对一生存者选择)
• rcs (Random Compensation Selection,随机补偿选择)
• rps (Random Permutation Selection,随机排列选择)
• rws (Roulette Wheel Selection,轮盘赌选择)
• sus (Stochastic Universal Sampling,随机抽样选择)
• tour (Tournament,锦标赛选择)
• urs (Uncommitted Random Selection,无约束随机选择)
5. 重组(包括交叉)
交叉是重组的一部分。
• recdis (离散重组)
• recint (中间重组)
• reclin (线性重组)
• recndx (正态分布交叉)
• recsbx (模拟二进制交叉)
• xovbd (二项式分布交叉)
• xovdp (两点交叉)
• xovexp (指数交叉)
• xovmp (多点交叉)
• xovox (顺序交叉)
• xovpmx (部分匹配交叉)
• xovsec (洗牌指数交叉)
• xovsh (洗牌交叉)
• xovsp (单点交叉)
• xovud (均匀分布交叉)
注意:所有重组算子都不会检查重组结果是否满足所设边界范围。下面的变异算子则是内置对边界范围的检查和修复。因此如果在进化算法中要单是使用重组算子,则需要调用“ea.boundfix”函数进行边界修复。详见“help(ea.boundfix)”。
在重组过程中,种群的前一半个体会与后一半个体的染色体按照个体顺序进行一一配对。这些重组算子可通过设置传入参数“Half”的值为True,来使得重组后只保留一半的个体,此时将保留上面所说的一一配对重组后的第一条染色体。
Geatpy2.2.2版本之后在进化算法框架中新增了面向对象的Recombination(重组算子接口),上述的低级重组算子均有与之对应的重组算子类来进行参数的设置及调用,这些新增的类命名为首字母大写的对应低级重组算子的名称:
• Recdis (离散重组算子类)
• Recint (中间重组算子类)
• Reclin (线性重组算子类)
• Recndx (正态分布交叉算子类)
• Recsbx (模拟二进制交叉算子类)
• Xovbd (二项式分布交叉算子类)
• Xovdp (两点交叉算子类)
• Xovexp (指数交叉算子类)
• Xovmp (多点交叉算子类)
• Xovox (顺序交叉算子类)
• Xovpmx (部分匹配交叉算子类)
• Xovsec (洗牌指数交叉算子类)
• Xovsh (洗牌交叉算子类)
• Xovsp (单点交叉算子类)
• Xovud (均匀分布交叉算子类)
在调用某个重组算子时,可以直接调用低级重组算子进行重组;也可以利用高级重组算子recombine 通过指定低级重组算子的名称来调用低级重组算子进行重组,如recombine(’xovdp’, ...);也可以通过实例化重组算子类的对象,然后执行该对象的do()函数进行重组,例如:recOper = Xovdp(...),recOper.do(...)。具体结构详见这些类的源码。
6. 突变
mutate 是高级的突变函数,它调用下面的低级突变函数:
• mutbga (Mutation for Breeder Genetic Algorithm,Breeder GA 算法突变算子)
• mutbin (Mutation for Binary Chromosomes,二进制变异算子)
• mutde (Mutation for Differential Evolution,差分变异算子)
• mutgau (Gaussian Mutation,高斯突变算子)
• mutinv (Invertion Mutation,染色体片段逆转变异算子)
• mutmove (Mutation by Moving,染色体片段移位变异算子)
• mutpolyn (Polynomial Mutation,多项式变异)
• mutpp (Mutation of Permutation Chromosomes,排列编码变异算子)
• mutswap (Two Point Swapping Mutation,染色体两点互换变异算子)
• mutuni (Uniform Mutation,均匀变异算子)
注意:对于mutbga、mutde、mutgau、mutpolyn、mutuni,变异是先按实数值来变异,然后对于标记了是离散型变量进行四舍五入。因此结果往往会是浮点“float”类型的,此时如果要把这些离散值用作其他变量的下标,需要对其进行强制类型转换。
Geatpy2.2.2 版本之后在进化算法框架中新增了面向对象的Mutation(变异算子算子
接口),上述的低级变异算子均有与之对应的变异算子类来进行参数的设置及调用,这
些新增的类命名为首字母大写的对应低级变异算子的名称:
• Mutbga (Mutation for Breeder Genetic Algorithm,Breeder GA 算法突变算子类)
• Mutbin (Mutation for Binary Chromosomes,二进制变异算子类)
• Mutde (Mutation for Differential Evolution,差分变异算子类)
• Mutgau (Gaussian Mutation,高斯突变算子类)
• Mutinv (Invertion Mutation,染色体片段逆转变异算子类)
• Mutmove (Mutation by Moving,染色体片段移位变异算子类)
• Mutpolyn (Polynomial Mutation,多项式变异类)
• Mutpp (Mutation of Permutation Chromosomes,排列编码变异算子类)
• Mutswap (Two Point Swapping Mutation,染色体两点互换变异算子类)
• Mutuni (Uniform Mutation,均匀变异算子类)
在调用某个变异算子时,可以直接调用低级变异算子进行重组;也可以利用高级变异算子mutate 通过指定低级变异算子的名称来调用低级变异算子进行重组,如mutate(’mutgau’, ...);也可以通过实例化变异算子类的对象,然后执行该对象的do() 函数进行变异,例如:mutOper = Mutgau(...),mutOper.do(...)。具体结构详见这些类的源码。
7. 染色体解码
对于二进制/格雷编码的种群,我们要对其进行解码才能得到其表现型。
• bs2int (二进制/格雷码转整数)
• bs2real (二进制/格雷码转实数)
• bs2ri (二进制/格雷码转实整数)
8. 数据可视化
• moeaplot (多目标优化目标空间绘图函数)
• soeaplot (单目标优化绘图函数)
• trcplot (进化记录器绘图函数)
• varplot (决策变量绘图函数)
如果是在iPython 控制台中调用可视化绘图函数(例如使用Spyder 开发工具),一般图像会默认显示在控制台中。此时可以在控制台下执行%matplotlib 来设置把图像显示在一个独立窗口中。
9. 多目标相关
• awGA (适应性权重法多目标聚合函数)
• rwGA (随机权重法多目标聚合函数)
• ndsortDED (基于排除法的帕累托层级划分)
• ndsortESS (基于ENS_SS 的快速非支配层级划分)
• ndsortTNS (基于T_ENS 的快速非支配层级划分)
• crtgp (创建在单位超空间中均匀的网格点集)
• crtup (创建在单位超平面内均匀分布的点集)
• crowdis (拥挤距离计算)
• refgselect (利用参考点引导的个体选择)
• refselect (基于参考点的“入龛”个体选择)
10. 模板相关
• soea_DE_best_1_bin_templet (差分进化DE/best/1/bin 算法模板)
• soea_DE_best_1_L_templet (差分进化DE/best/1/L 算法模板)
• soea_DE_rand_1_bin_templet (差分进化DE/rand/1/bin 算法模板)
• soea_DE_rand_1_L_templet (差分进化DE/rand/1/L 算法模板)
• soea_DE_targetToBest_1_bin_templet (差分进化DE/targetToBest/1/bin 算法模板)
• soea_DE_targetToBest_1_L_templet (差分进化DE/targetToBest/1/L 算法模板)
• soea_ES_1_plus_1_templet ((1+1) 进化策略模板)
• soea_EGA_templet (精英保留的遗传算法模板)
• soea_SEGA_templet (增强精英保留的遗传算法模板)
• soea_SGA_templet (最简单、最经典的遗传算法模板)
• soea_GGAP_SGA_templet (带代沟的简单遗传算法模板)
• soea_studGA_templet (种马遗传算法模板)
• soea_steady_GA_templet (稳态遗传算法模板)
• soea_psy_EGA_templet (精英保留的多染色体遗传算法模板)
• soea_psy_SEGA_templet (增强精英保留的多染色体遗传算法模板)
• soea_psy_SGA_templet (最简单、最经典的多染色体遗传算法模板)
• soea_psy_GGAP_SGA_templet (带代沟的多染色体简单遗传算法模板)
• soea_psy_studGA_templet (多染色体种马遗传算法模板)
• soea_psy_steady_GA_templet (多染色体稳态遗传算法模板)
• moea_awGA_templet (基于awGA 算法的多目标进化算法模板)
• moea_NSGA2_DE_templet (基于NSGA-II-DE 算法的多目标进化算法模板)
• moea_NSGA2_archive_templet (带全局存档的多目标进化NSGA-II 算法模板)
• moea_NSGA2_templet (基于NSGA-II 算法的多目标进化算法模板)
• moea_NSGA3_DE_templet (基于NSGA-III-DE 算法的多目标进化算法模板)
• moea_NSGA3_templet (基于NSGA-III 算法的多目标进化算法模板)
• moea_RVEA_templet (基于RVEA 算法的多目标进化算法模板)
• moea_RVEA_RES_templet (基于带参考点再生策略的RVEA 算法的多目标进化算法
模板)
• moea_psy_awGA_templet (基于awGA 算法的多染色体多目标进化算法模板)
• moea_psy_NSGA2_archive_templet (带全局存档的多染色体多目标进化NSGA-II 算
法模板)
• moea_psy_NSGA2_templet (基于NSGA-II 算法的多染色体多目标进化算法模板)
• moea_psy_NSGA3_templet (基于NSGA-III 算法的多染色体多目标进化算法模板)
• moea_psy_RVEA_templet (基于RVEA 算法的多染色体多目标进化算法模板)
• moea_psy_RVEA_RES_templet (基于带参考点再生策略的多染色体RVEA 算法的多
目标进化算法模板)
详细API文档可以在Python控制台中执行help(模块名)查看。
下面讲一下Geatpy中重要的数据结构:
Geatpy的大部分数据都是存储在numpy的array数组里的,numpy中另外还有matrix的矩阵类型,但我们不使用它,于是本文档默认array就是存储“矩阵”(也可以存储一维向量,接下来会谈到)。其中有一些细节需要特别注意:numpy的array在表示行向量时会有2种不同的结构,一种是1行n列的矩阵,它是二维的;一种是纯粹的一维行向量。因此,在Geatpy教程中会严格区分这两种概念,我们称前者为“行矩阵”,后者为“行向量”。Geatpy中不会使用超过二维的array。
例如有一个行向量x,其值为[1 2 3 4 5 6],那么,用print(x.shape)输出其规格,可以得到(6,),若x是行矩阵而不是行向量,那么x的规格就变成是(1,6)而不再是(6,)。
在numpy的array类型中,实际上没有“列向量”的概念。所谓“向量”是指一维的,但用numpy的array表示列向量时,它实际上是二维的,只不过只有1列。我们不纠结于这个细节,统一仍用“列向量”来称呼这种只有1列的矩阵。
在编程中,如果对numpy的array感到疑惑,你可以用"print(变量.shape)"语句来输出其维度信息,以确定其准确的维度。
- 种群染色体的数据结构:
Geatpy中,种群染色体是一个numpy的array类型的二维矩阵,一般用Chrom命名,每一行对应一个个体的一条染色体。若要采用多染色体,则可以创建多个相关联的Chrom即可。默认一个Chrom的一行对应的只有一条染色体。
我们一般把种群的规模(即种群的个体数)用Nind命名;把种群个体的染色体长度用$Lind$命名,则Chrom的结构如下所示:

- 种群表现型的数据结构:
种群表现型的数据结构跟种群染色体基本一致,也是numpy的array类型。我们一般用Phen来命名。它是种群染色体矩阵Chrom经过解码后得到的基因表现型矩阵,每一行对应一个个体,每一列对应一个决策变量。若用Nvar表示变量的个数,则种群表现型矩阵Phen的结构如下图:

Phen的值与采用的解码方式有关。Geatpy提供二进制/格雷码编码转十进制整数或实数的解码方式。另外,在Geatpy也可以使用不需要解码的“实值编码”种群,这种种群的染色体的每一位就对应着决策变量的实际值,即这种编码方式下Phen等价Chrom。
这里需要注意的是:我们可以用不同的方式去解码一个种群染色体,得到的结果往往是不同的。
- 目标函数值的数据结构:
Geatpy采用numpy的array类型矩阵来存储种群的目标函数值。一般命名为ObjV,每一行对应每一个个体,因此它拥有与Chrom相同的行数;每一列对应一个目标函数,因此对于单目标函数,ObjV会只有1列;而对于多目标函数,ObjV会有多列。例如ObjV 是一个二元函数值矩阵:

其第一列就代表目标函数f1的目标函数值,第二列代表目标函数f2的目标函数值。
- 个体适应度的数据结构:
Geatpy采用列向量来存储种群个体适应度。一般命名为FitnV,它同样是numpy的array类型,每一行对应种群矩阵的每一个个体。因此它拥有与Chrom相同的行数。

Geatpy中的适应度遵循“最小适应度为0”的约定。
- 个体违反约束程度的数据结构
Geatpy采用Numpy array类型的矩阵CV(Constraint Violation Value)来存储种群个体违反各个约束条件的程度。一般命名为$CV$,它的每一行对应种群的每一个个体,因此它拥有与$Chrom$相同的行数;每一列对应一个约束条件,因此若有一个约束条件,那么CV矩阵就会只有一列,如有多个约束条件,CV矩阵就会有多列。如果设有$num$个约束,则CV矩阵的结构如下图所示:

CV矩阵的某个元素若小于或等于0,则表示该元素对应的个体满足对应的约束条件。若大于0,则表示违反约束条件,在大于0的条件下值越大,该个体违反该约束的程度就越高。Geatpy提供两种处理约束条件的方法,一种是罚函数法,另一种是可行性法则。在使用可行性法则处理约束条件时,需要用到CV矩阵。具体用法可详见后面两章中关于使用可行性法则来处理约束条件的相关说明。
- 译码矩阵的数据结构:
Geatpy使用译码矩阵(俗称区域描述器)来描述种群染色体的特征,如染色体中的每一位元素所表达的决策变量的范围、是否包含范围的边界、采用二进制还是格雷码、是否使用对数刻度、染色体解码后所代表的决策变量的是连续型变量还是离散型变量等等。
在只使用工具箱的库函数而不使用Geatpy提供的面向对象的进化算法框架时,译码矩阵可以单独使用。若采用Geatpy提供的面向对象的进化算法框架时,译码矩阵可以与一个存储着种群染色体编码方式的字符串Encoding来配合使用。目前Geatpy中有三种Encoding,分别为:
• ’BG’ (二进制/格雷码)
• ’RI’ (实整数编码,即实数和整数的混合编码)
• ’P’ (排列编码,即染色体每一位的元素都是互异的)
这里有个小小的归类值得注意:'RI'和'P'编码的染色体都不需要解码,染色体上的每一位本身就代表着决策变量的真实值,因此“实整数编码”和“排列编码”可统称为“实值编码”。
1) 对于$Encoding = 'BG'$的种群,使用8行$n$列的矩阵FieldD来作为译码矩阵,$n$是染色体所表达的决策变量个数。FieldD的结构如下:

lens, lb, ub, codes, scales, lbin, ubin, varTypes 均为长度等于决策变量个数的行向
量。
其中,lens 包含染色体的每个子染色体的长度。sum(lens) 等于染色体长度。
lb 和ub 分别代表每个决策变量的上界和下界。
codes 指明染色体子串用的是二进制编码还是格雷编码。codes[i] = 0 表示第i 个变量使用的是标准二进制编码;codes[i] = 1 表示使用格雷编码。
scales 指明每个子串用的是算术刻度还是对数刻度。scales[i] = 0 为算术刻度,scales[i] = 1 为对数刻度。对数刻度可以用于变量的范围较大而且不确定的情况,对于大范围的参数边界,对数刻度让搜索可用较少的位数,从而减少了遗传算法的计算量。(注意:当某个变量是对数刻度时,其取值范围中不能有0,即要么上下界都大于0,要么上下界都小于0。)
lbin 和ubin 指明了变量是否包含其范围的边界。0 表示不包含边界;1 表示包含边界。
varTypes 指明了决策变量的类型,元素为0 表示对应位置的决策变量是连续型变量;1 表示对应的是离散型变量。
例如:

它表示待解码的种群染色体矩阵Chrom解码后可以表示成3个决策变量,每个决策变量的取值范围分别是[1,10], [2,9], [3,15]。其中第一第二个变量采用的是二进制编码,第三个变量采用的是格雷编码,且第一、第三个决策变量为连续型变量;第二个为离散型遍历。若Chrom为:

则可以执行以下语句进行解码:
import geatpy as ea
Phen = ea.bs2ri(Chrom, FieldD)解码后得到的种群表现型矩阵为:
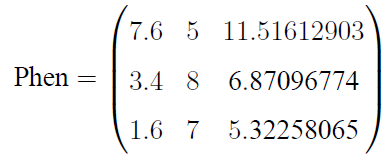
2) 对于实值编码(即前面所说的不需要解码的编码方式) 的种群,使用3 行n 列的矩阵FieldDR 来作为译码矩阵,n 是染色体所表达的控制变量个数。FieldDR 的结构如下:

这种结构的译码矩阵适用于Encoding 为’RI’(实整数编码) 和’P’(排列编码) 的种群染色体的解码。其中’P’(排列编码) 的译码矩阵会稍微有一点特殊之处:它要求FieldDR的第一行所有元素都相等,第二行所有元素也都相等,且第三行元素均为1(这是因为排列编码本身变量是离散的)。此时若记FieldDR 有Lind 列(即染色体长度为Lind),则要求上界- 下界+ 1 ≥ Lind。例如:

它若是作为排列编码种群的译码矩阵,则表示种群染色体是由集合{2,3,4,5,6,7,8,9,10}中任意抽取7 个数出来的全排列,比如Chrom 为:

上面的FieldD 和FieldDR 都是numpy 的array 类型,可统称为“Field”。可以直接用代码创建,例如:
import numpy as np
FieldDR=np.array([[-3, -4, 0, 2],
[ 2, 3, 2, 2],
[ 0, 0, 0, 0]])也可以用Geatpy 内置的crtfld 函数来方便地快速生成区域描述器,其详细用法可执行help(crtfld) 或查看API 文档。
- 进化追踪器的数据结构:
在使用Geatpy 进行进化算法编程时,常常建立一个进化追踪器(如pop_trace) 来记录种群在进化的过程中各代的最优个体,尤其是采用无精英保留机制时,进化追踪器帮助我们记录种群在进化的“历史长河”中产生过的最优个体。待进化完成后,再从进化追踪器中挑选出“历史最优”的个体。这种进化记录器有多种,其中一种是numpy 的array 类型的,结构如下:
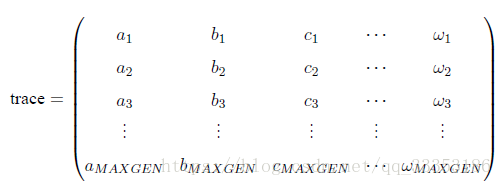
其中MAXGEN 是种群进化的代数。trace 的每一列代表不同的指标,比如第一列记录各代种群的最佳目标函数值,第二列记录各代种群的平均目标函数值……trace 的每一行对应每一代,如第一行代表第一代,第二行代表第二代……
另外一种进化记录器是一个列表,列表中的每一个元素都是一个拥有相同数据类型的数据。比如在Geatpy 的面向对象进化算法框架中的pop_trace,它是一个列表,列表中的每一个元素都是历代的种群对象。
- 种群结构
1) Population 类
在Geatpy 提供的面向对象进化算法框架中,种群类(Population) 是一个存储着与种群个体相关信息的类。它有以下基本属性:
sizes : int - 种群规模,即种群的个体数目。
Lind : int - 种群染色体长度。
Encoding : str - 染色体编码方式。
Field : array - 译码矩阵,可以是FieldD或FieldDR。
Chrom : array - 种群染色体矩阵,每一行对应一个个体的一条染色体。
ObjV : array - 种群目标函数值矩阵。
FitnV : array - 种群个体适应度列向量。
CV : array - 种群个体违反约束条件程度的矩阵。
Phen : array - 种群表现型矩阵。
可以直接对种群对象进行提取个体、个体合并等操作,比如pop1和pop2是两个种群对象,则通过语句“pop3 = pop1 + pop2”,即可把两个种群的个体合并,得到一个新的种群。在合并的过程中,实际上是把种群的各个属性进行合并,然后用合并的数据来生成一个新的种群(详见Population.py)。又比如执行语句“pop3 = pop1[[0]]”,可以把种群的第0号个体抽取出来,得到一个新的只有一个个体的种群对象pop3。值得注意的是,种群的这种个体抽取操作要求下标必须为列表或是Numpy array类型的行向量,不能是标量(详见Population.py)。
易错注意:在Geatpy中,必要地对种群对象的这些成员属性进行合法性检查是必要的,但过多的检查会在一定程度上降低框架的性能。其中最容易使得种群对象成员属性出现异常的地方在于目标函数值矩阵ObjV以及CV矩阵的生成。在Geatpy中,ObjV和CV是在Problem问题类的目标函数接口aimFunc()中计算生成的,无论中间过程它们具体是如何计算的,计算得到的结果必须满足:ObjV和CV都是Numpy array类型矩阵,且行数等于种群的个体数目。ObjV的每一行对应一个个体,每一列对应一个优化目标。CV矩阵的每一行也是对应一个个体,而每一列对应一个约束条件。根据Geatpy数据结构可知,种群对象中的Chrom、ObjV、FitnV、CV和Phen都是Numpy array类型的行数等于种群规模sizes的矩阵,即这些成员属性的每一行都跟种群的每一个个体是一一对应的。Geatpy框架在运行过程中所抛出大多数异常都是由于这些变量不合法所致。此时可以使用“shape”来输出变量的维度信息,比如:
print(pop.sizes)
print(pop.Chrom.shape)
print(pop.ObjV.shape)
print(pop.CV.shape)其中pop为一个种群对象。
2)PsyPopulation类
PsyPopulation类是Population的子类,它提供Population类所不支持的多染色体混合编码。它有以下基本属性:
sizes : int - 种群规模,即种群的个体数目。
ChromNum : int - 染色体的数目,即每个个体有多少条染色体。
Linds : list<int> - 存储种群各染色体长度的列表。
Encodings : list<string> - 存储各染色体编码方式的列表。
Fields : list<array> - 存储各染色体对应的译码矩阵的列表。
Chroms : list<array> - 存储种群各染色体矩阵的列表。
ObjV : array - 种群目标函数值矩阵。
FitnV : array - 种群个体适应度列向量。
CV : array - 种群个体违反约束条件程度的矩阵。
Phen : array - 种群表现型矩阵。
可见PsyPopulation类基本与Population类一样,不同之处是采用Linds、Encodings、Fields和Chroms分别存储多个Lind、Encoding、Field和Chrom。
PsyPopulation类的对象往往与带“psy”字样的进化算法模板配合使用,以实现多染色体混合编码的进化优化。
在后面我将详细阐述如何使用Geatpy提供的面向对象进化算法框架的算法模板来实现使用进化算法求解问题。
欢迎继续跟进,感谢!














 311
311











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









