









文章转自:
作者:黄树嘉
链接:https://www.jianshu.com/p/8cd6ef12b2e2
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
[注] 本文同时发于泛基因fungenomics公众号和我的个人博客。
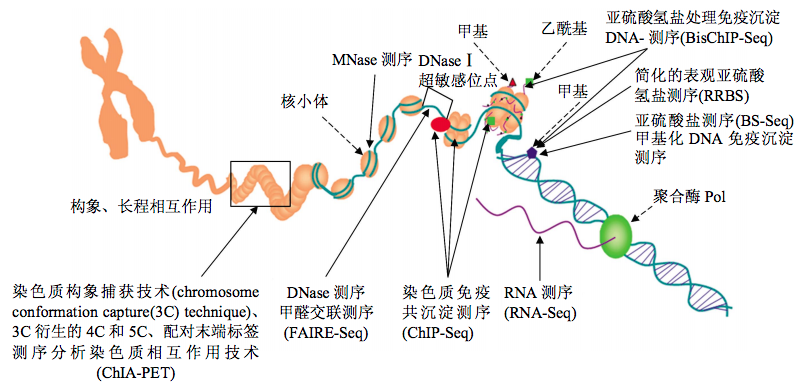
Deep Learning,现在几乎到处都能看到它的应用。看!紧随DeepBind,在基因组学应用中又来了一个DeepSEA——这是一个适用于表观遗传研究和应用的工具,它只从DNA序列出发,并没用其他有关于表观研究的实验或者测序技术,通过直接输入fasta sequence,vcf或者bed文件,就可以预测转录因子结合位点(Transcription factors binding site), DNase I超敏感位点(DNase I hypersensitive sites)和组蛋白靶点(histone marks),这么多年来,这样的做法还是头一回。下面这张示意图展示的是各个主要的表观修饰在染色体中的位置和相关实验测定技术。
EPI
为什么要有这么个东西呢?
众所周知,人类基因组上绝大部分的序列都是非编码序列——不直接编码蛋白质的序列,这些序列在很长的一段时间里都被误解为所谓的“垃圾DNA”!但其实它们各自都有着独特的作用——调控着机体的正常运作,只是要想正确地理解它们确实不是一个容易的事情。DeepSEA想要干的就是尝试从序列的基础功能预测着手去解决这么一个难题。
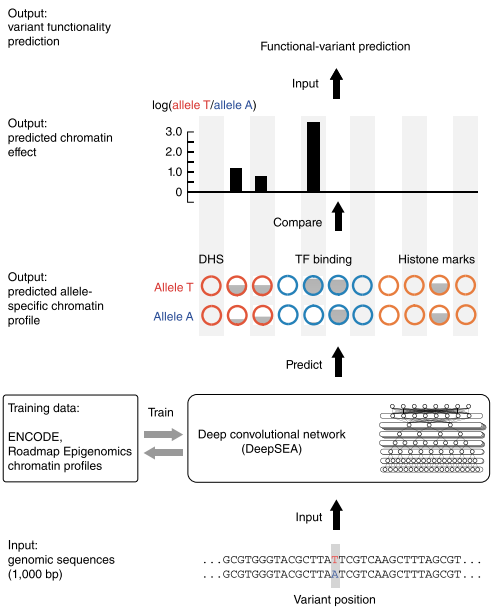
deepsea
它先通过学习大量已知的染色质修饰数据——主要来自于ENCODE和Roadmap Epigenomics等大型项目,经过不断的训练,学习到了许多种在非编码区域中序列调控的序列模式或者说是序列特征(注意是序列模式,不是功能模式),之后,便可以通过这些模式和特征去预测序列上单碱基的突变会如何影响染色质的修饰功能。从发表的文章来看,其精确程度是目前所有方案中最高也是在同等数据下最有效的了。
DeepSEA 在Nature Method的原文http://www.nature.com/nmeth/journal/v12/n10/full/nmeth.3547.html
更赞的是它的代码和相关训练数据都一起公开在网站上:http://deepsea.princeton.edu/ 可以尝试玩起来了。
下面我:
在GitHub上找了个代码跑一下:
https://github.com/jiawei6636/Bioinfor-DeepSEA
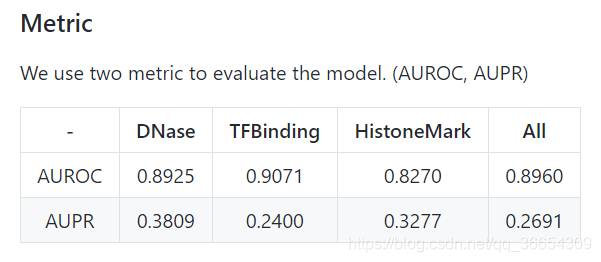

















 1157
1157

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









