






教程的完整输入文件在demos/tutorials/中。
文章目录
一、Minimizer
1.介绍
蛋白质不是静态结构,而是在其构象中经历波动,并以状态集合的形式存在。
蛋白质构象库中的每一个构象都与能量有关,其中一些构象具有高能量,一些具有低能量。在分子建模中,通常需要找到这个能量函数的全局最小值(代表最低能量构象)。然而,考虑到需要搜索的巨大能源景观,这是一个非常困难的任务,所以我们将满足于下一个最好的事情:局部最小值。
minimizaton是在给定初始结构的构象和能量的情况下,求能量函数中最接近的局部极小值的一种技术。
Rosetta有一个核心算法,叫做minimizer,它解决了将结构移动到其最近的局部能量最小值的问题。它执行了梯度下降最小化的几种变化之一,以找到能量函数中最近的局部最小值。minimizer可以使用许多不同的最小化算法,但本质上,所有的最小化算法都选择一个向量作为下降方向,沿着这个向量前进,直到能量停止下降(“linear search”),然后选择一个新的方向并重复。在本教程中,我们将使用 lbfgs/_armijo/_nonmonotone,这是一个多步骤算法,只需要调用一次就可以达到函数的局部最小值(而不是调用重复迭代来达到收敛)。
一般来说,minimizatoin是确定的,不像依赖于蒙特卡罗搜索的方法。重复多次极小化轨迹通常没有什么好处;总体生成不应该有多样性。
也就是说,minimization对于不同平台,甚至不同编译器之间的微小数值精度差异非常敏感。
许多Rosetta模块调用minimizer,但最简单的方法之一是使用minimizer可执行程序。
2.目标
在本教程中,您将学习以三种不同的方式使用许多最小化算法之一,首先通过允许结构中的所有残基在最小化期间移动,然后通过使用两种方法,只允许改变自由度(DOFs)的子集。具体来说,我们将:
- 学习通过命令行运行 score 和 minimize可执行文件。
- 创建和利用一个约束文件,以防止CA原子的移动发生在结构的一部分。
- 创建并利用一个movemap来控制最小化过程中允许移动的自由度。
- 比较和分析每种最小化类型的输出文件和结构。
3. How-To: Minimize
分析输入结构
获得一个蛋白质3hon.pdb构象的最初分数,在命令行输入:
$> <path_to_Rosetta>/main/source/bin/score.default.linuxgccrelease -s 3hon.pdb
这个命令会输出一个包含此构象的Rosetta分数的文件default.sc 。在这个文件中,我们可以看到:

这个文件的第一行告诉我们每一列所对应的分数项。
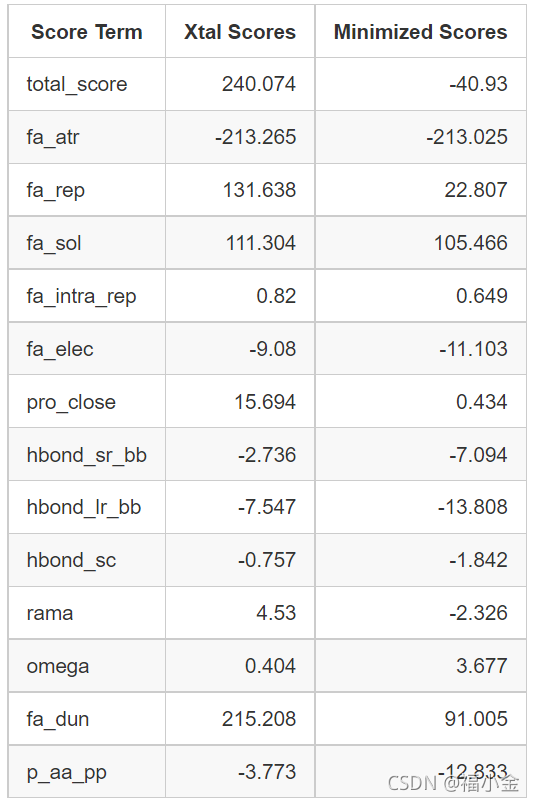
在这个文件的第二行,我们看到晶体构象的Rosetta总分240.074, 它有一个特别高repulsive的分数, fa_rep, 这意味着结构中原子之间有冲突, 还有一个高dunbrack分数, fa_dun, 意味着这个结构中许多旋转异构体的概率较低。
让我们尝试使用minimizer来修复这些问题。
设置flags文件
首先我们需要详细说明如何运行minimizer。打开 minimizer_flags 文件,并分析它的内容:
-s 3hon.pdb
-run:min_type lbfgs_armijo_nonmonotone
-run:min_tolerance 0.001
第一个符号 s,指定了输入文件,在本例中是3hon的晶体结构。
第二个符号 run:min_type,指定了要使用的最小化算法的类型,在本例中是 lbfgs_armijo_nonmonotone.
( lbfgs_armijo_nonmonotone是默认的算法,所以这行可以省略)
第三个符号 run:min_tolerance,指定了最小化算法的收敛阈值。对于函数最小化,Rosetta至少有两种“公差”,“常规”公差(没有更好的名字)和绝对公差。“常规”公差是被最小化的函数值的分数公差;即0.01的公差意味着最小函数值很可能在真实局部最小值的1%以内。而绝对公差不考虑当前函数值;即绝对容差为0.01,意味着所找到的最小函数值与真实局部最小值之间的距离最多为0.01REU。minimizer默认使用“常规”分数公差。通常,将分数公差设置为0.01是非常宽松的,因此建议指定一个小于0.01的公差设置。因此,在本教程中,我们将公差设置为0.001。
运行minimization命令
在你的终端窗口,输入以下命令运行minimization可执行文件:
$> <path_to_Rosetta>/main/source/bin/minimize.default.linuxgccrelease @minimizer_flags
如果这个可执行文件运行时没有错误,并且终端输出以类似这样的内容结束,
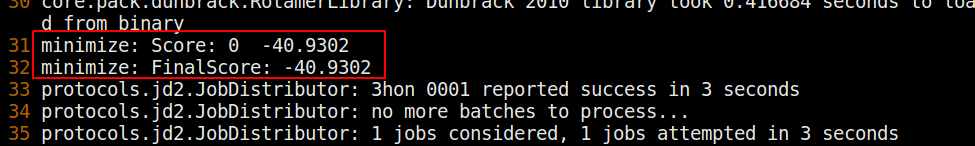
这样就成功地最小化了输入结构。检查以确保生成了一个score.sc 文件和一个3hon_0001.pdb 输出结构。现在我们来分析输出分数和结构。
分析输出
score.sc 文件包含最小化结构的新分数。打开这个文件并和最初的晶体结构分数进行比较:
minimizaton是如何影响结构的呢?打开3hon_0001.pdb 看看什么发生了改变。
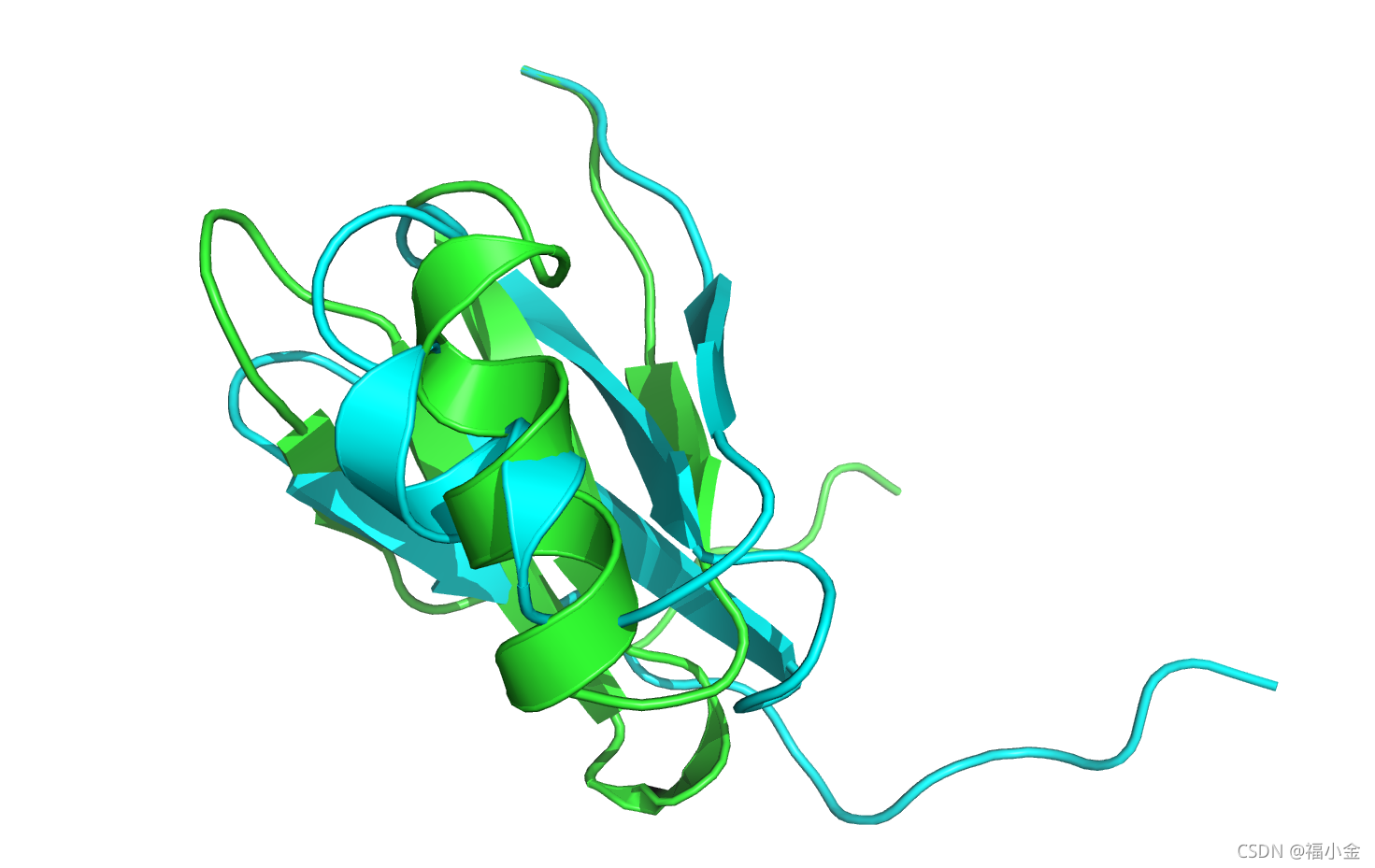
最小化结构(绿色)已经偏离了与天然结构(蓝色)的比对,所以首先比对这两个结构。如果你使用的是PyMOL,输入align 3hon_0001, 3hon, 回车。
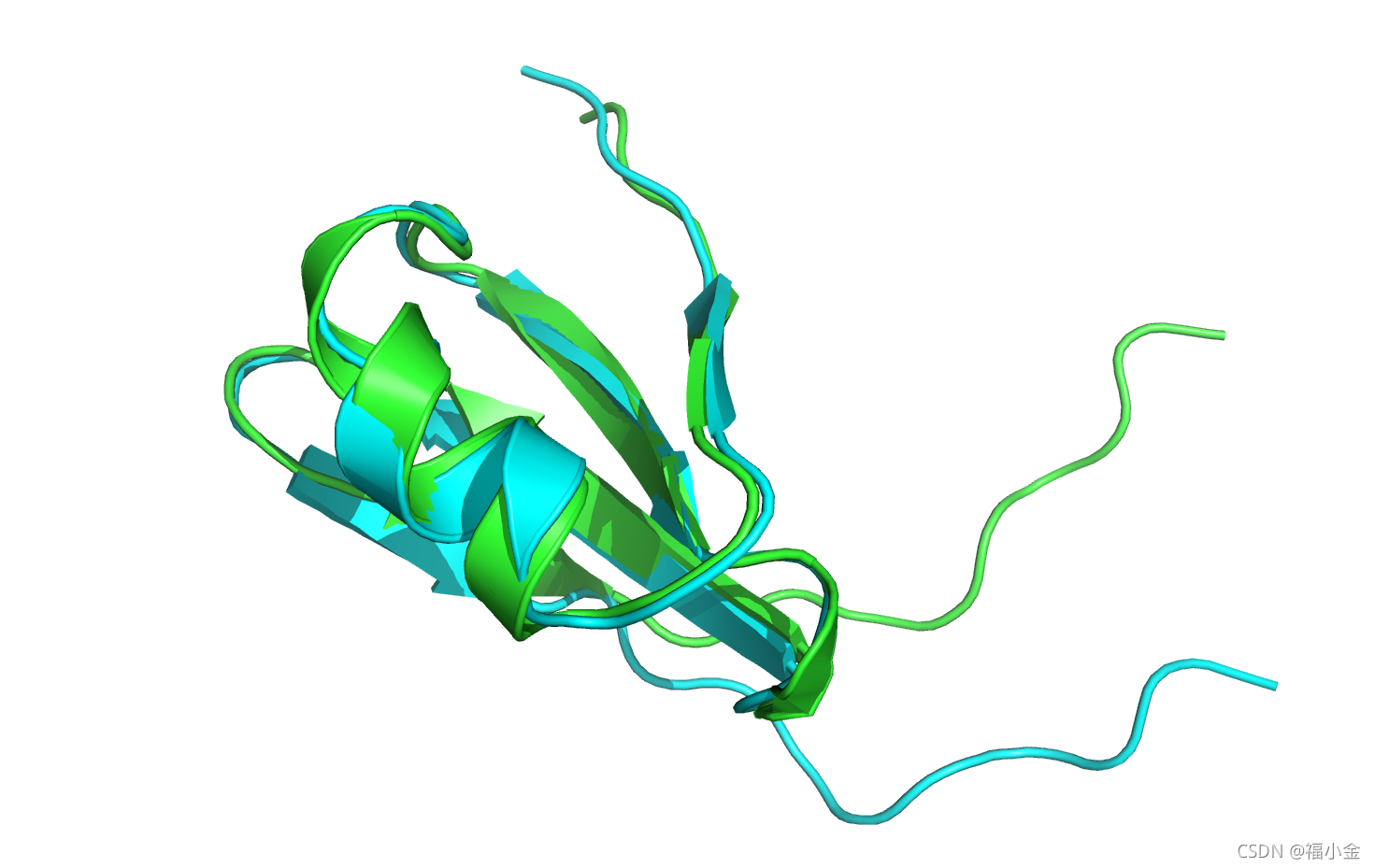
现在结构已经对齐了,注意到loop区的最后9个残基已经从原来的构象明显地移动了。此外,大多数最小化的旋转异构体不再是它们的天然构象。
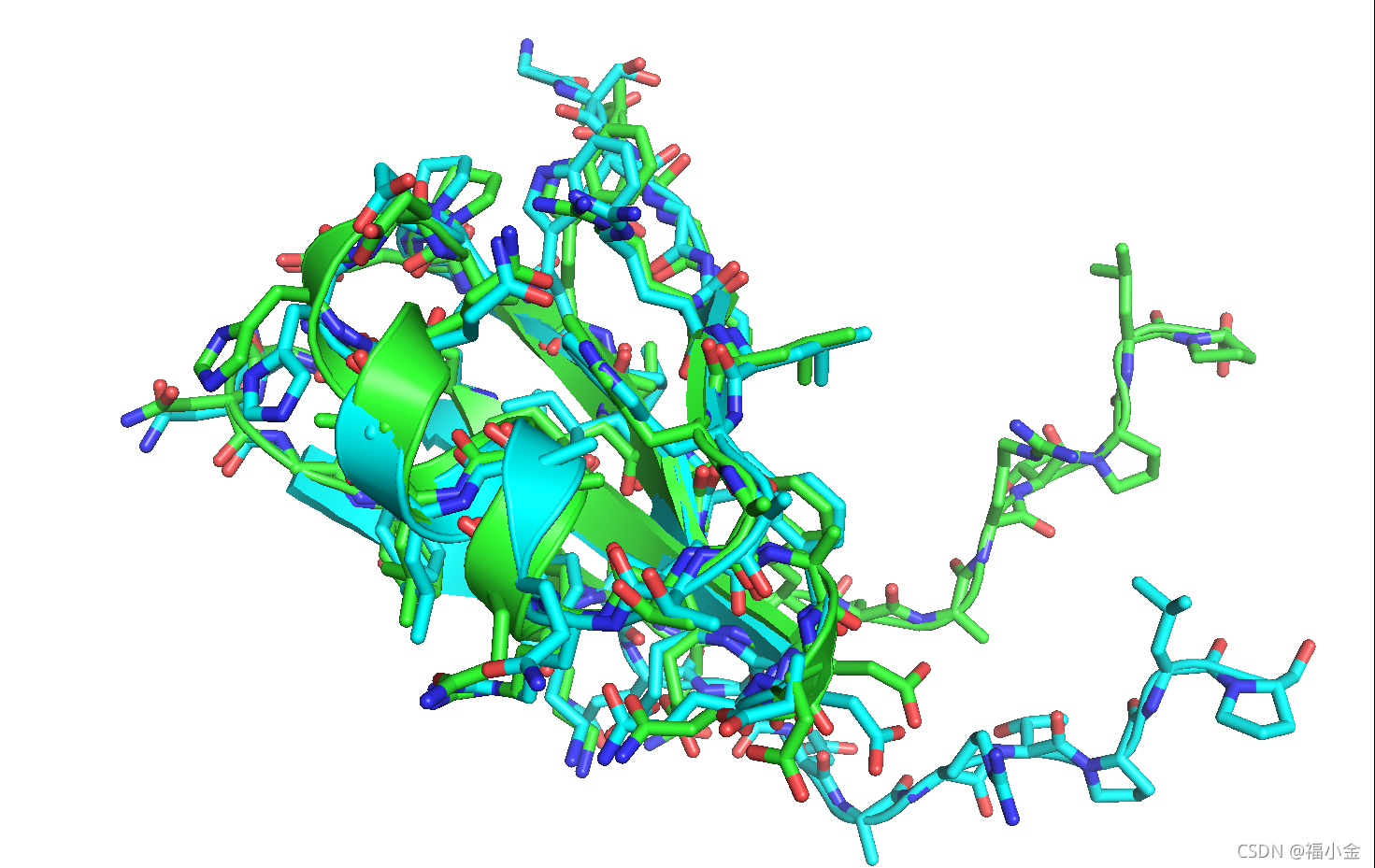
有时在起始构象中允许如此大的运动是不可取的。为此,我们可以使用带约束的minimization来最小化我们的输入结构,在这个结构中,某些原子的运动将被分数函数惩罚。
4. How-To: Minimization with Constraints
在本节教程中,我们将使用与之前相同的晶体结构,3hon.pdb,但这次我们将对九个c端尾部残基的主链重原子应用harmonic coordinate constraints (csts)。你可以把这些约束想象成“松紧带”或“橡皮筋”,在最小化轨道中轻轻地把每个主链重原子拉回到原来的位置。实际上,约束是添加到总能量函数中的额外的人工势能(一系列惩罚偏离输入坐标的harmonic 势能)。
设置flags文件和约束文件
我们需要向flags文件添加额外的选项来告诉Rosetta读取那些指定如何将原子固定在适当位置的约束。此外,我们需要告诉Rosetta在其使用的能量函数中添加额外的约束势能。为此,我们在weight文件中为约束分数项设置非零权。新的flags文件,minwithcsts_flags,看起来像这样:
-s 3hon.pdb
-run:min_type lbfgs_armijo_nonmonotone
-run:min_toleranace 0.001
-constraints:cst_file cstfile
-score:weights ref2015_cst
-out:suffix _minwithcsts
符号 constraints:cst_file 指定了使用的约束文件的名称。
符号score:weights 指定了能量函数使用哪一组weights。在本例中,我们指定了ref2015_cst 权重文件,这是Rosetta数据库中预定义的权重文件,除了添加了约束项 chainbreak、坐标约束、原子对约束、角度约束、二面体约束和res_type_constraint之外,它与ref2015(默认的scorefunction)相同, 每个权重都是1.0。
选项 out:suffix 通过在新的输出文件名的末尾添加后缀“_minwithcsts”来防止我们覆盖之前的输出。
运行minimization命令
以和之前同样的方式运行minimization可执行文件,但现在使用了新的flags文件:
$> <path_to_Rosetta>/main/source/bin/minimize.default.linuxgccrelease @minwithcsts_flags
这一次,在日志输出中查找 core. score .constraints 跟踪器中的几行。它们看起来应该与此类似(尽管不同平台之间可能存在数值差异):
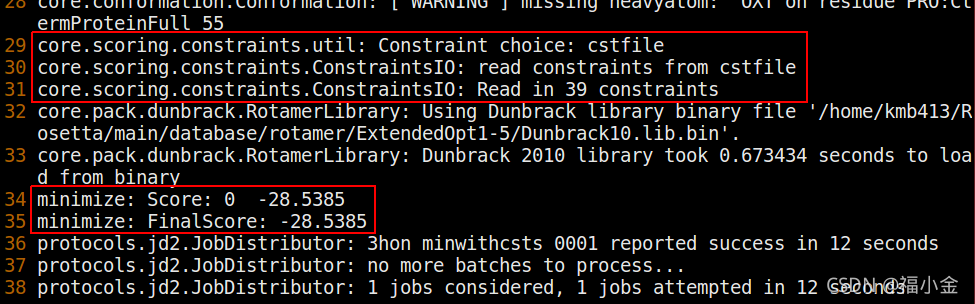
这些行表示我们的约束条件已被读取并应用到pose上。如果可执行文件在没有错误的情况下结束并生成一个3hon_minwithcsts_0001.pdb文件,以及 score-minwithcsts.sc 文件,那么你就成功地运行了带有坐标约束的最小化。
分析输出
和之前一样,score-minwithcsts.sc 文件包含受约束的最小化结构的分数。打开此文件,并将其能量与晶体结构以及无约束最小化生成的结构进行比较:
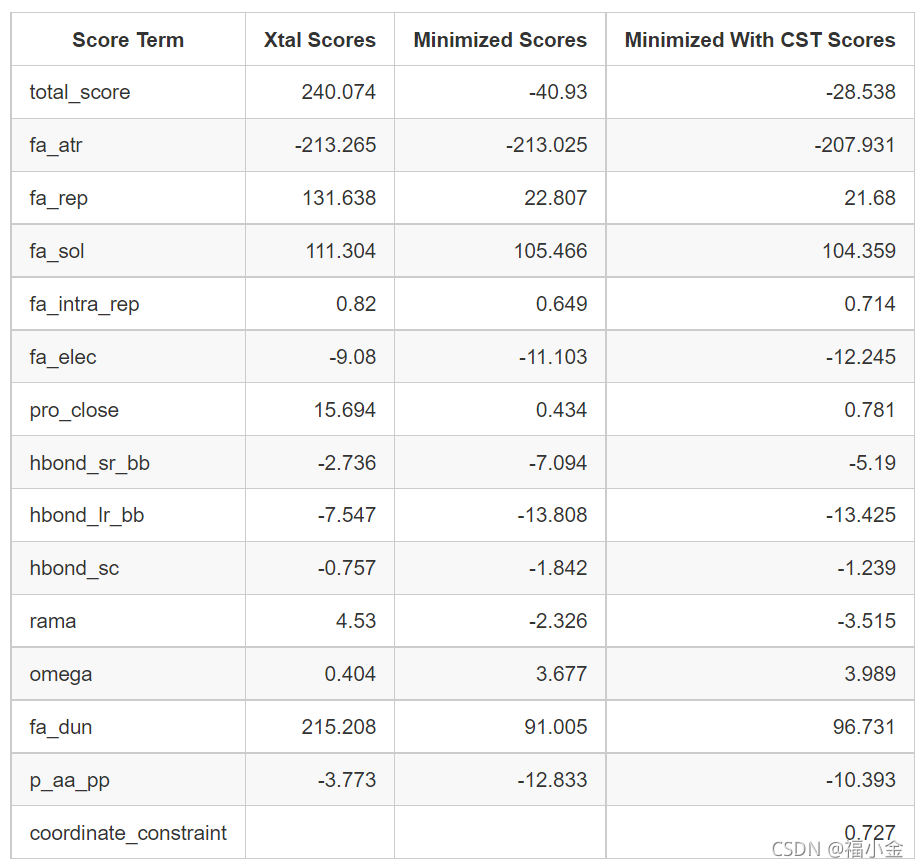
同样,大多数来自带约束的最小化结构的新分数都低于晶体结构的分数。还要注意,对于新最小化的结构,在eneray项列表中增加了坐标约束项。
比较最小化的结构和带约束的最小化结构,我们看到总能量的增加主要是由fa_atr和fa_dun项的差异引起的。这有两个来源。首先,我们看到坐标约束项本身为分数增加了一个小的正值,因为主链重原子从它们的起始坐标稍微移动了一点。第二,因为这次的结构没有被允许移动得那么远,小的冲突和缺陷不会完全地自行解决,导致分数比以前稍微高一些。因此,在保持结构接近晶体结构和深入挖掘到最近的局部能量最小之间存在一种权衡。
现在,让我们看看最小化的csts结构,看看它与前面的结构相比如何。
打开 3hon_minwithcsts_0001.pdb 文件并和它的晶体结构比较,
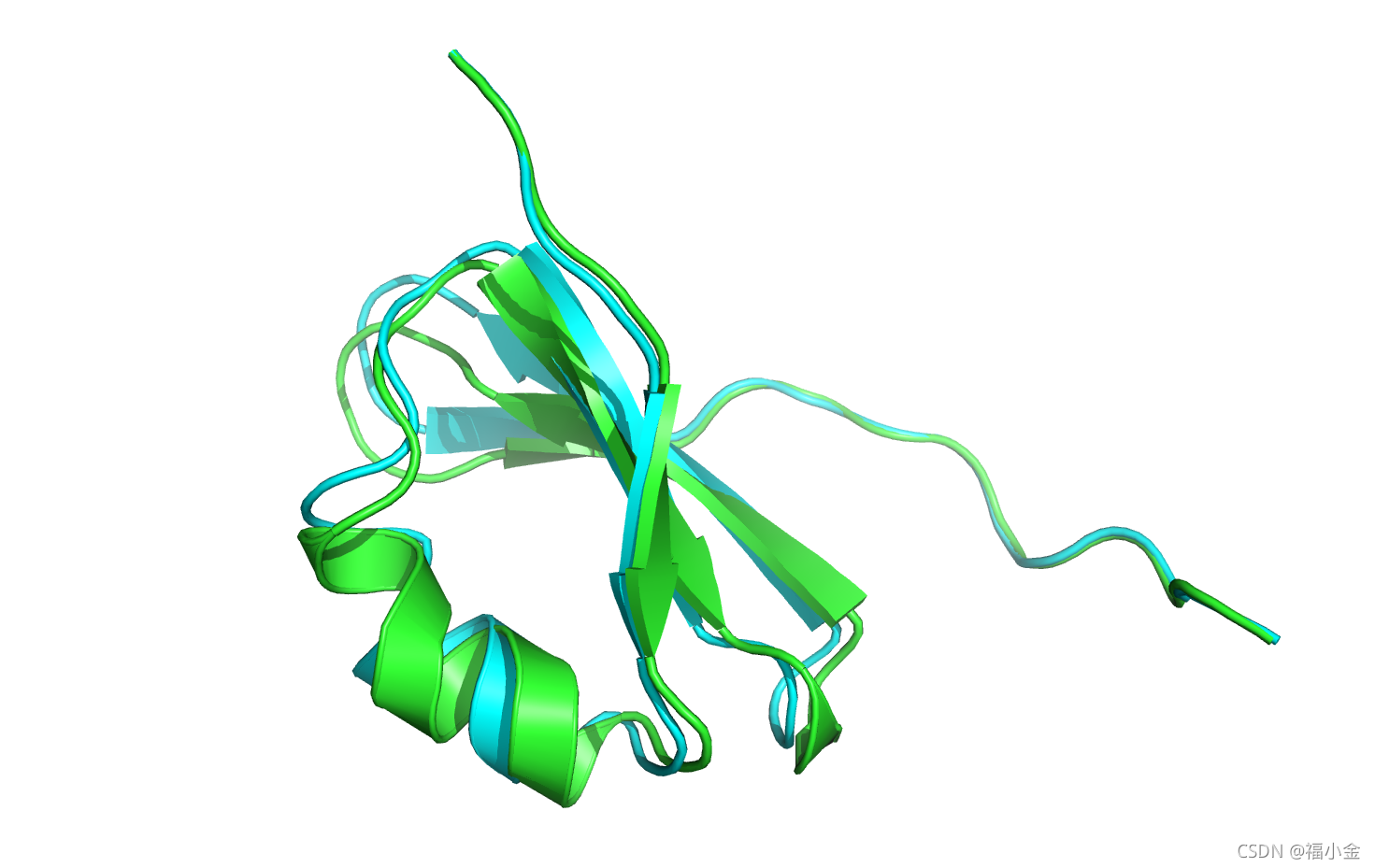
我们立刻就能看到9个c端CA原子的位置几乎没有移动。
在某些情况下,用户可能希望在最小化过程中防止某些残基的内部几何形状移动,而不是原子的XYZ坐标。为了禁止主链phi/psi角和/或侧链chi角的移动,我们可以为minimizer提供一个MoveMap,指定哪些自由度可以改变。
5. How-To: Minimization with a MoveMap
The MoveMap
movemap是Rosetta最小化中的一个重要概念。它允许用户控制最小化过程中哪些自由度可以改变,哪些自由度是固定的。例如,人们可能不希望在建模应用程序中移动高度保守的侧链,或者希望在设计应用程序中保留某些交互。某些调用Rosetta minimizer的协议接受用户定义的movemap文件。
在minimizer的背景下,movemap允许用户指定在能量函数最小化期间是否允许主链(BB)扭转角(在α-氨基酸残基的情况下是phi和psi)和/或侧链扭转角(CHI)的移动。此外,如果输入结构有多个链,那么movemap还可以指定是否允许在不同链之间进行刚体移动。
注意:即使残基的主链和侧链扭转运动在一个movemap中被关闭,它相对于其他残基的相对位置仍然可能根据折叠树中残基上游的运动而改变。
关于FoldTree的说明
foldtree是另一个与最小化相关的重要概念。虽然foldtree的操作将在另一个教程中有更详细的介绍,但我们将在这里给出一个简单的概念概述。当最小化时,自由度会发生变化,这可能会对结构产生模糊的影响。例如,如果我把残基5的扭转从-60改变到-50,会发生几件事: 残基1到4可能保持不变所有从5开始的残基都可能移动; 残基1到4可能移动,5可能保持不变;残基5中的原子可能移动,但蛋白质中的其余原子保持不变,这意味着蛋白质的键几何结构会严重扭曲;或者是三者的某种组合。foldtree建立了pose中残基之间的层次关系,从而定义了变化自由度的影响。每个残差有一个惟一的父残基(除了一个根残差之外),每个残差可以有一个或多个子残差。(循环依赖是禁止的——例如,如果残基3是残基4的父级,那么残基4就不能是残基3的父级。)残基自由度的变化会传播到残基的所有孩子,但不影响父级。在一个典型的pose下,大多数残基是通过键连接联系起来的,而两个残基可以通过一个穿过空间的刚体变换联系起来,称为jump。如果存在两个独立的链(没有键连接它们),则必须至少有一个将第一个的残基与第二个的残基关联起来的jump。
如上所述,调用minimizer的用户,特别是使用MoveMap的用户,应该注意foldtree。默认的foldtree以A链的残基1为根,残基2为1的子链,残基3为2的子链,等等,并跳转到任何附加链的第一个残差。任何可移动的自由度都会影响foldtree中其下游的残基自由度,即使残基自由度是由MoveMap固定的。作为一个具体的例子,让我们考虑一个带有默认foldtree的20个残基的pose,使用一个MoveMap来防止残基8到20的主链自由度的移动。能量最小化会改变残基1到7的主链自由度,在这个过程中,残基8到20在空间中摇摆(尽管在这种情况下,残基8到20不能相互相对移动)。
movemap文件格式的描述
movemap文件中的每一行都识别一个jump、残基或残基范围,后面跟着允许的自由度。这些对象可以如下指定:
RESIDUE <#> <BB/CHI/BBCHI/NO> # a single residue <#> followed by a single option (pick one of the four)
RESIDUE <#1> <#2> <BB/CHI/BBCHI/NO> # a range of residues from <#1> to <#2> followed by a single option (again, pick one)
JUMP <#> <YES/NO> # a jump <#> followed by a single option
例如,
RESIDUE 28 BB # allows BB movements at residue 28
RESIDUE 32 48 BBCHI # allows BB and sidechain CHI movements from residue 32 to residue 48
JUMP 1 YES # allows rigid-body movements between the structures separated by jump 1
也可以用 * 符号选择所有残基或所有jumps:
RESIDUE * CHI # allows sidechain movements at all residues
JUMP * YES # allows rigid body movements between all structures separated by jumps
如果一个残基出现了不止一次,则文件中的最后一次出现决定了移动(例如,movemap行是不可交换的,不像用于控制包装器的TaskOperations——请参阅打包教程)。例如,这里指定的移动地图…
RESIDUE * CHI
RESIDUE * BB
…将只允许所有残基的主链(BB)移动,将不允许所有残基的侧链(CHI)移动,这可能不是用户的意思。
注意:如果在movemap中没有指定残基或jump,它将恢复到默认行为,这是协议特定的。在许多但不是所有情况下,默认行为是所有扭转自由度(bb, chi)和所有刚体自由度(jumps)都是可移动的。当有疑问时,最好明确说明用户请求的行为。
设置flags文件和movemap文件
对于下一个minimization步骤,我们需要在flags文件中添加一个选项,告诉Rosetta读取movemap文件。新的flags文件 minwithmm_flags 标志的内容应该是这样的:
-s 3hon.pdb
-run:min_type lbfgs_armijo_nonmonotone
-run:min_toleranace 0.001
-movemap movemapfile
-out:suffix _minwithmm
符号 movemap 指定了应用到pose的movemap文件。
我们的movemap文件 movemapfile (下面展开)告诉minimizer首先将所有主干(BB)和侧链(CHI)的扭转角度设置为可移动。然后,它将残基47到55(在pose中的编号)进行固定。
RESIDUE * BBCHI
RESIDUE 47 55 NO
运行minimization命令
以和之前同样的方式运行minimization可执行文件,但现在使用了新的flags文件:
$> <path_to_Rosetta>/main/source/bin/minimize.default.linuxgccrelease @minwithmm_flags
日志输出应该与第一个最小化协议相似,只是分数有所不同。在运行此命令之后你也应该有一个3hon_minwithmm_0001.pdb 文件和一个score_minwithmm.sc 文件。
分析输出
比较分数:
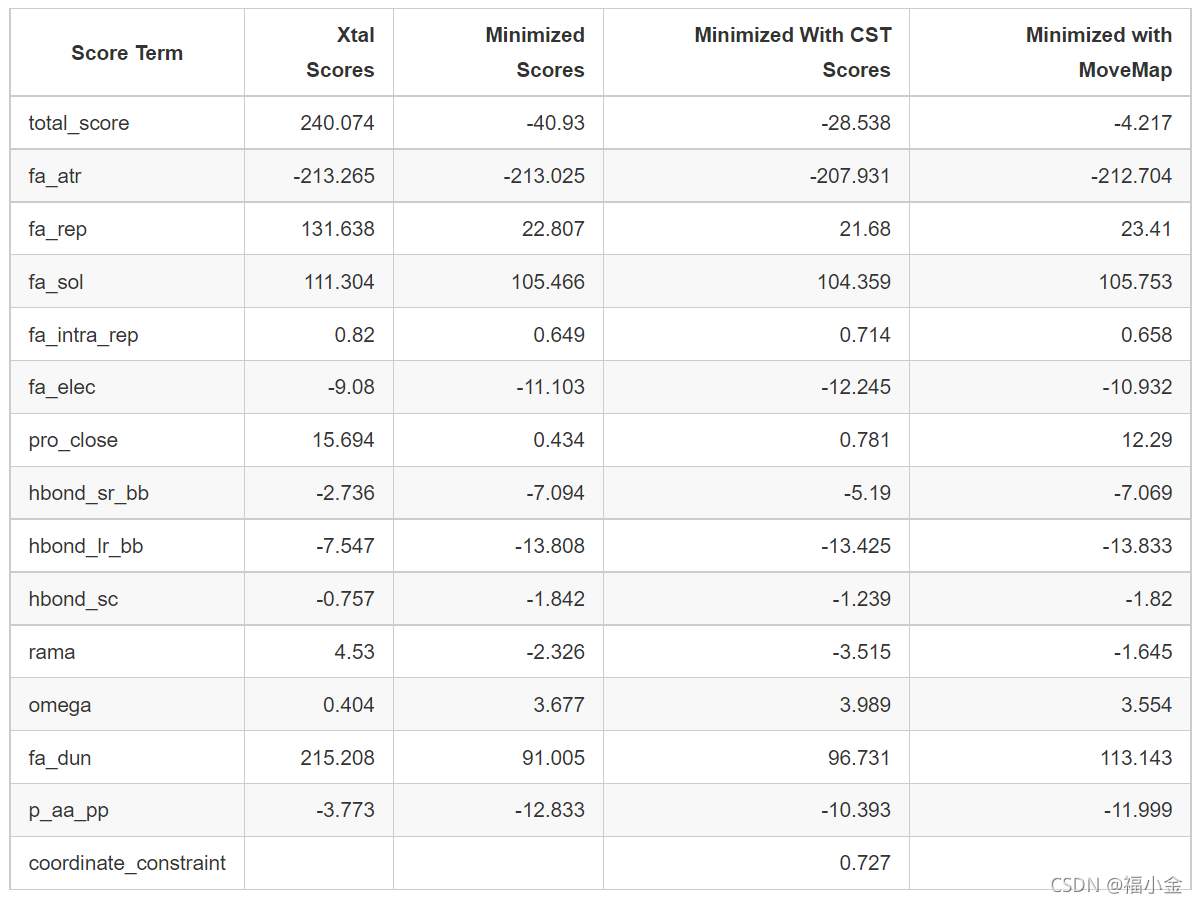
带有movemap的最小化结构的总得分仍然低于晶体结构,但在最小化结构中具有最高的能量,这主要是由于与脯氨酸环闭合相关的能量pro_close项和fa_dun项的差异造成的。
让我们打开带有movemap的最小化结构,并将其与晶体结构进行视觉上的比较。
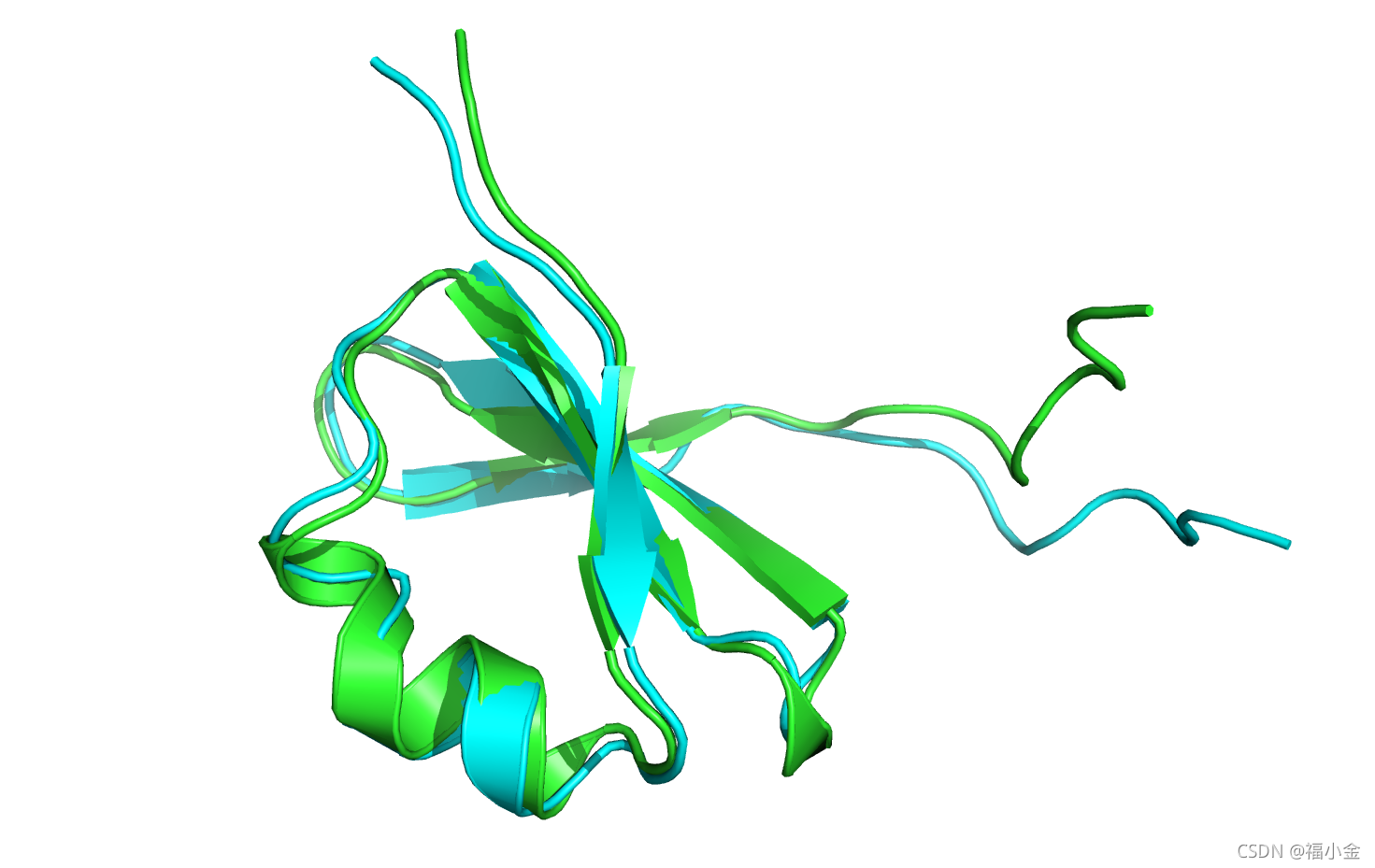
将带有movemap的最小化结构与晶体结构整体对齐后,9个c端残基似乎发生了移动。然而,当我们只将9个c端残基彼此对齐时,很明显minimizer并没有改变主链或侧链的角度:
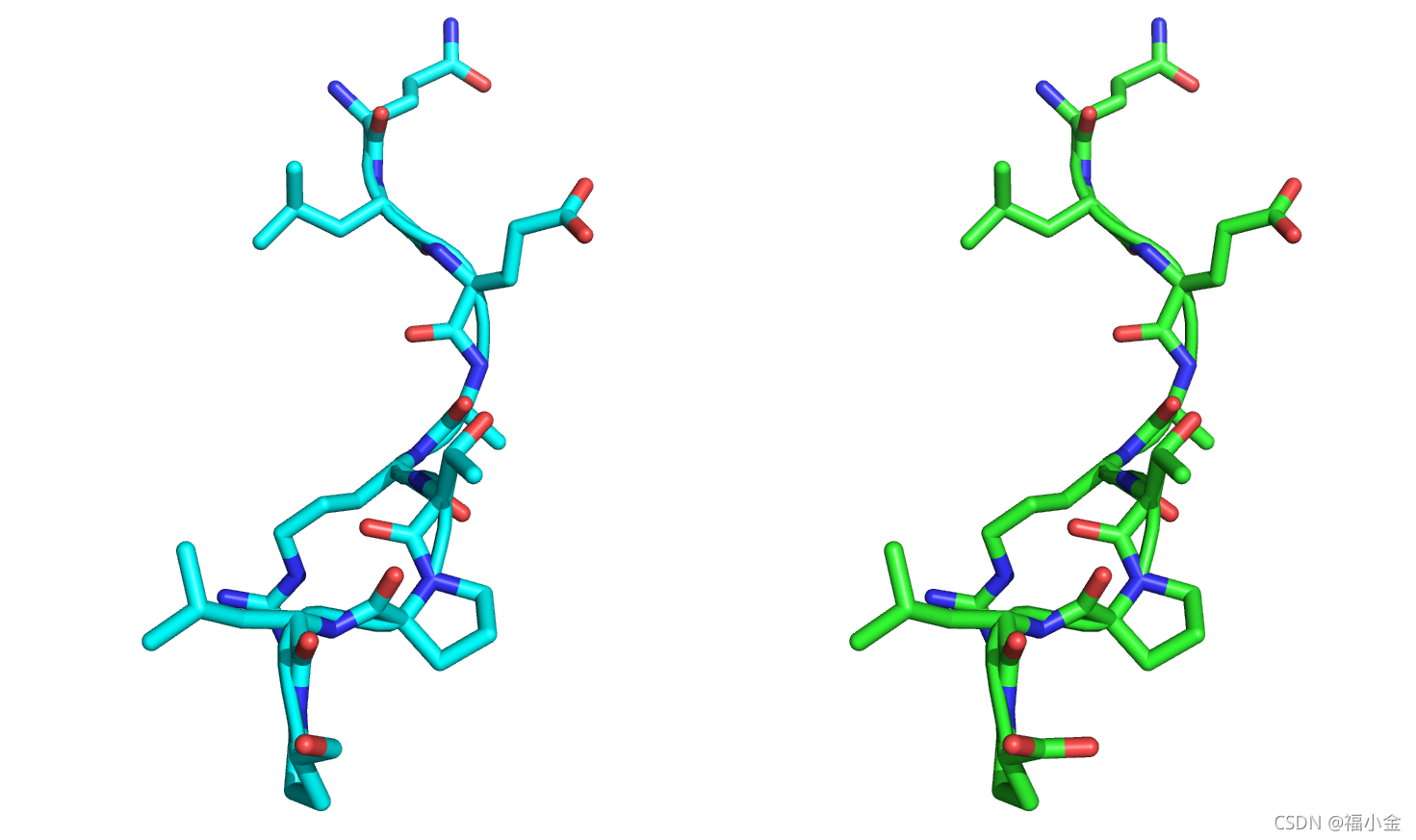
需要重申的一点很重要: movemap可以防止内部几何图形的改变,但不一定是残基的全局位置。
6. 总结
总结在本教程中,我们学习了如何以三种方式运行minimize 可执行文件:
- 使用默认行为,它允许所有的主链和侧链角移动,
- 通过对输入结构应用坐标约束,禁止原子子集的全局移动,
- 通过应用MoveMap,禁止主链和侧链扭转角的局部移动。

















 377
377

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









