







小罗碎碎念
肿瘤微环境研究的挑战与机遇
在肿瘤研究领域,肿瘤微环境(TME)的细胞组成分析至关重要。
传统的分析方法,如流式细胞术、单细胞RNA测序(scRNA-seq)和免疫组化,虽各有优势,但在成本、细胞类型覆盖范围和提取效率等方面存在明显局限。
批量RNA测序虽具有成本效益,然而癌细胞基因表达谱(GEP)的高度变异性,以及现有计算方法难以充分考虑细胞类型内的异质性、确定最佳基因列表和处理高维数据等问题,使得准确推断肿瘤细胞组成面临重重困难,这也为医学AI研究在该领域的应用带来了挑战与机遇。
DeSide方法的创新突破
针对上述难题,一种名为DeSide的统一深度学习方法应运而生。它整合了来自多种实体肿瘤和癌细胞系的scRNA-seq数据集,运用创新的采样和数据过滤技术,生成与实际肿瘤高度相似的合成肿瘤GEP。

在模型架构上,DeSide采用独特的深度神经网络(DNN),结合生物通路信息作为额外特征,通过特定的激活函数和网络结构设计,有效克服了癌细胞GEP变异带来的干扰,能够准确估计16种细胞类型在实体肿瘤中的比例。
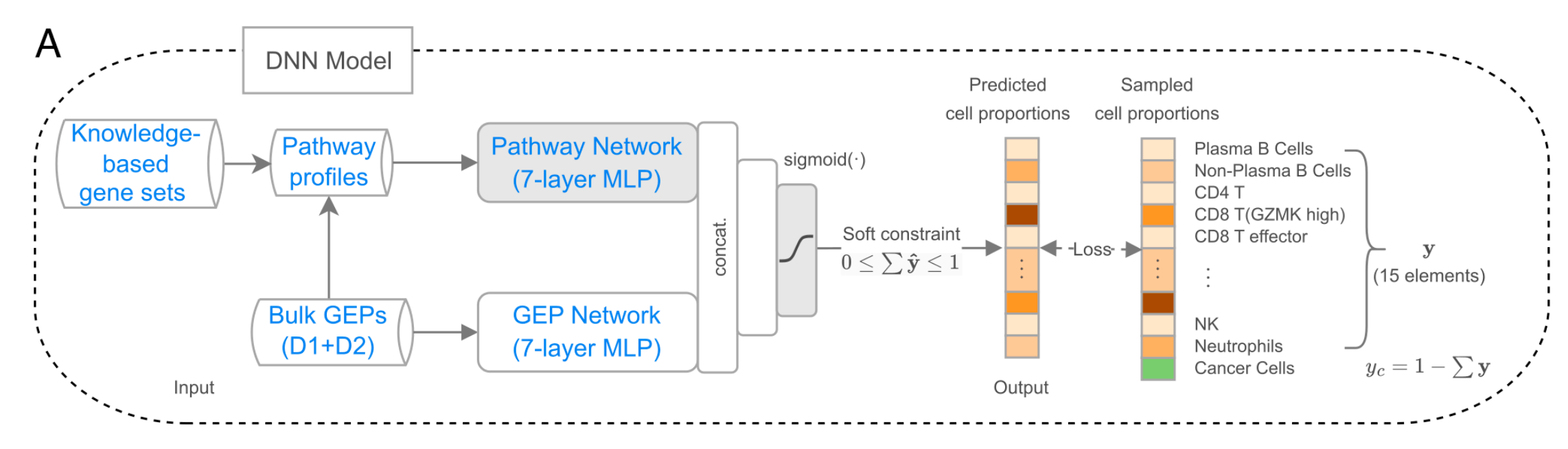
实验结果显示,DeSide在预测肿瘤纯度和细胞组成方面,显著优于现有方法,并且在多个外部数据集上展现出良好的泛化能力。
DeSide的应用价值与未来展望
DeSide在医学AI研究中具有重要的应用价值。一方面,它能够通过准确估计细胞比例,实现对癌症患者的有效分层,为临床预后评估提供有力支持。
研究发现,基于DeSide预测的细胞比例,可识别出多种对患者预后有显著影响的细胞类型及细胞类型组合,这有助于深入理解肿瘤生物学机制,挖掘潜在的治疗靶点。
另一方面,尽管DeSide目前存在数据归一化和未考虑细胞空间分布等局限性,但随着更多scRNA-seq数据的积累和技术的不断改进,其在肿瘤研究和临床应用中的潜力巨大,有望为医学AI在肿瘤微环境分析领域开辟新的方向,推动精准医疗的发展。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量52,000+,交流群总成员1100+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
如需获取推文中提及的各种资料,欢迎加入我的知识星球!
一、文献概述
- 研究背景
- 肿瘤微环境复杂性:实体肿瘤由癌细胞和肿瘤微环境(TME)中的多种细胞组成,TME细胞的存在和数量与患者预后相关,精确评估肿瘤细胞组成对临床治疗意义重大。
- 现有分析方法的局限性:常用的细胞组成分析方法如流式细胞术、单细胞RNA测序(scRNA-seq)和免疫组化存在成本高、细胞类型覆盖有限或提取效率低等问题。批量RNA测序虽具成本效益,但癌细胞基因表达谱(GEP)的高度变异性以及现有计算方法的缺陷,使得准确推断细胞比例面临挑战。
- DeSide方法
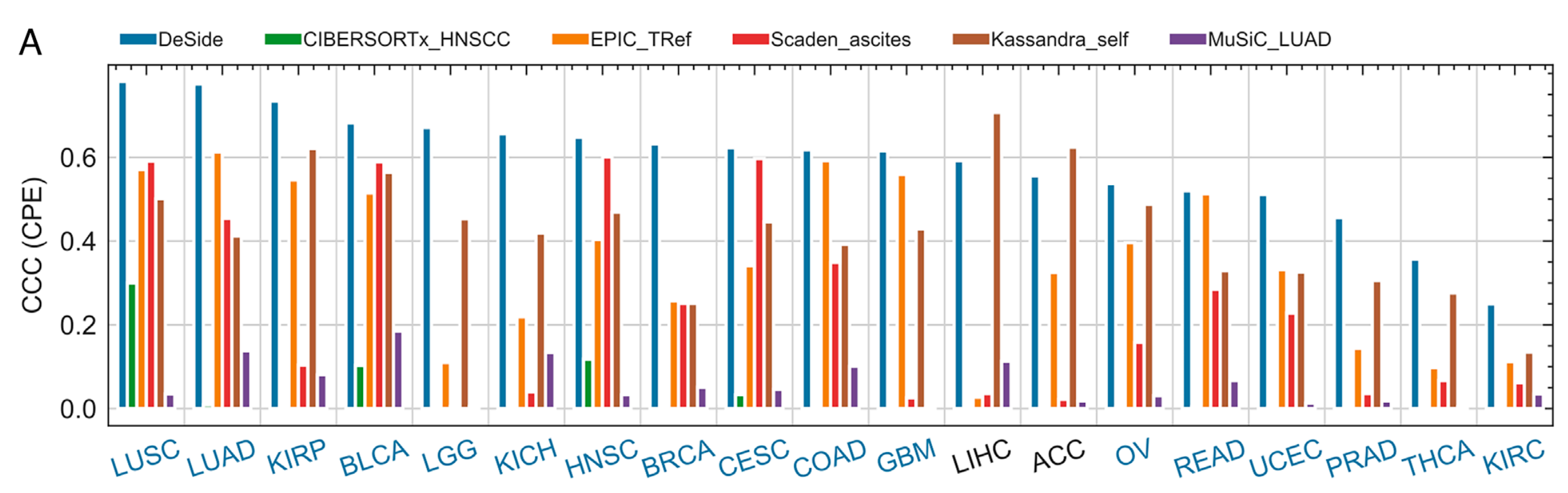
- DNN架构:采用两个全连接神经网络,即路径网络和GEP网络,分别从路径概况和GEP中提取信息。输出层使用sigmoid函数,对预测比例施加软约束,先预测非癌细胞比例,再计算癌细胞比例。
- 合成GEP策略:整合多个scRNA-seq数据集,创建包含16种细胞类型、每种类型10,000个单细胞类型GEP的平衡数据集S1。采用段采样生成细胞比例矩阵,实施GEP级和基因级过滤,提高合成肿瘤GEP与TCGA数据的相似性。
- 实验结果
- 训练集对模型性能的影响:不同训练集显著影响DNN模型性能,“段采样无过滤”(D2)生成的训练集覆盖更广泛的GEP空间,包含D1的训练集能提高与真实肿瘤的相似性,D1+D2组合训练集效果最佳。
- 与其他算法的性能比较:在预测19种肿瘤类型的癌细胞比例时,DeSide在17种肿瘤类型上的一致性相关系数(CCC)最高,优于其他基于特征基因的方法。在多个额外数据集上,DeSide对多种细胞类型的预测准确性也很高。
- 患者分层:基于DeSide预测的细胞比例,可将患者有效分层。在8种肿瘤类型中,多种细胞类型比例能显著区分患者的总生存期(OS),细胞类型组合分析发现了更多有意义的细胞相互作用。
- 研究讨论
- 优势:DeSide能准确估计细胞比例,在多种癌症类型中表现出色,且所需训练样本少,可用于GEP反卷积,有助于揭示肿瘤免疫和患者预后的关系。
- 局限:批量和单细胞RNA测序数据的归一化方法不同,可能引入偏差;DeSide未考虑细胞的空间分布信息。
- 材料和方法
- 合成肿瘤的GEP:生成D0、D1、D2三个数据集用于训练,对数据集S0进行处理生成S1,计算合成肿瘤的GEP并进行过滤,最终保留约9,000个基因。
- DNN模型:详细介绍了模型架构、路径概况计算方法、模型训练和测试等相关内容。
- 数据、材料和软件可用性:DeSide基于Python和TensorFlow实现,代码和数据集开源,可从GitHub、Zenodo等平台获取。
比较项目
| 比较项目 | 详情 |
|---|---|
| 训练集 | D0:随机采样无过滤;D1:段采样后过滤;D2:段采样无过滤;D1+D2:D1和D2组合 |
| 测试集 | T0 - T2:分别与D0 - D2生成方法相同;T3:由sctGEP生成 |
| 性能评估指标 | CCC、Pearson相关性(r)、均方根误差(RMSE)、平均绝对误差(MAE) |
| 数据集 | TCGA:19种肿瘤类型批量RNA-seq数据;GSE184398:12种实体肿瘤类型数据;SC_GBM、SC_HNSCC、SC_OV:特定癌症类型数据 |
| 细胞类型 | 预测16种独立细胞类型和7种合并细胞类型 |
| 患者分层指标 | Cox p值、log-rank p值、q值、风险比(HR) |
关键问题
- DeSide与其他细胞组成分析算法相比,优势体现在哪些方面?
- DeSide在预测肿瘤纯度和细胞组成方面准确性更高,在19种肿瘤类型中的17种上,其预测癌细胞比例与癌症纯度(CPE值)的一致性相关系数(CCC)最高。
- 它所需训练样本仅20万个,约为当前最先进方法Kassandra的1%。而且,DeSide能有效捕捉多种实体肿瘤的共同结构,泛化能力强,在不同数据集上对多种细胞类型的预测表现都很出色。
- DeSide在患者分层方面有哪些重要发现?
- 基于DeSide预测的细胞比例,在8种肿瘤类型中,多种细胞类型如癌症相关成纤维细胞(CAFs)、肌成纤维细胞、CD4 T细胞、肥大细胞等的比例,可有效将患者分层为总生存期(OS)显著不同的两组。
- 细胞类型组合分析发现,某些细胞类型组合能更有效地分层患者,如头颈部鳞状细胞癌(HNSCC)患者中,高CD4+ T细胞和低巨噬细胞比例的组合,与最佳OS相关,且危险比(HR)是仅使用CD4+ T细胞比例的两倍。
- DeSide目前存在哪些局限性?
- 批量和单细胞RNA测序数据的归一化方法不同,可能在预测细胞比例时引入系统偏差。
- DeSide目前未考虑细胞在肿瘤内的空间分布信息,而细胞的空间分布会影响患者预后和治疗反应。
二、DeSide概述
(A) 深度神经网络(DNN)模型
该模型由两个全连接神经网络组成:通路网络和基因表达谱(GEP)网络,每个网络都是一个七层的多层感知器(MLP),并共享一个输出层。
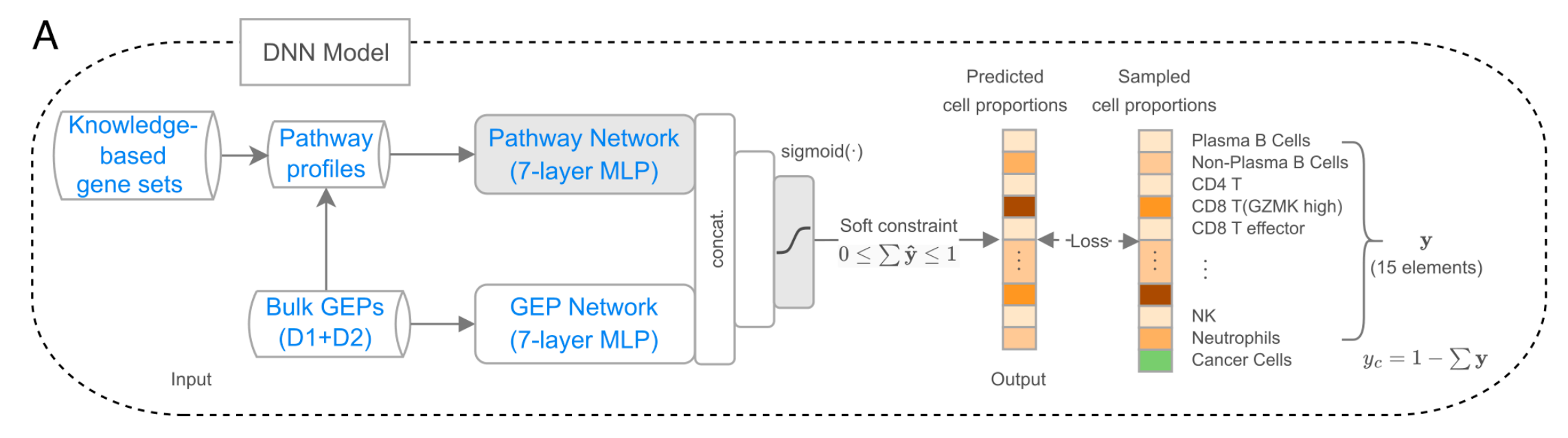
它们分别以肿瘤组织的通路概况和基因表达谱作为输入。经过处理后,它们的输出(来自第五个隐藏层)被连接起来,以从肿瘤组织的基因表达谱预测15种非恶性细胞类型的细胞比例(y)。
然后,通过从1中减去y的总和来计算癌细胞比例(yc),输出层使用sigmoid函数作为激活函数,并对输出总和应用软约束,即总和大于(或等于)0且小于(或等于)1。
(B) 肿瘤组织合成工作流程
此工作流程包括单细胞数据集整合、细胞比例生成和肿瘤组织基因表达谱模拟。
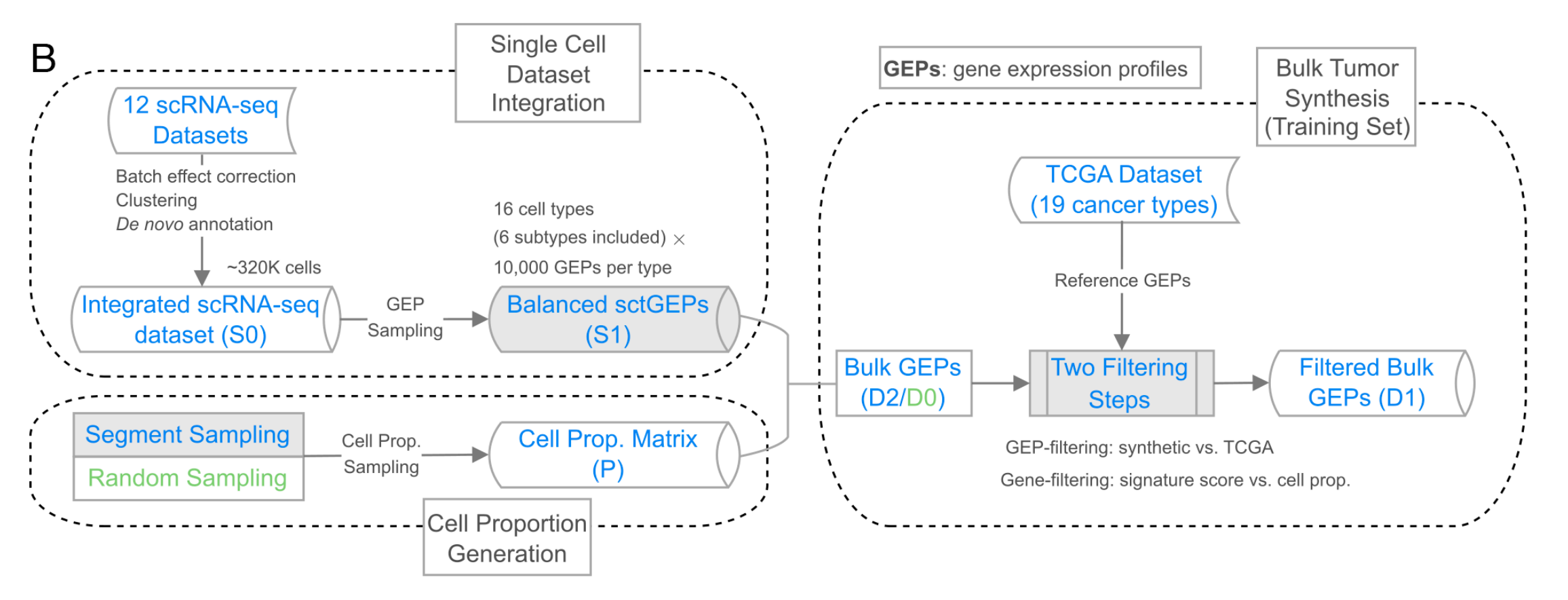
- 单细胞数据集整合:整合12个单细胞RNA测序(scRNA-seq)数据集,经批次效应校正、聚类和重新注释后,得到包含约32万个细胞的整合scRNA-seq数据集S0。从S0中采样基因表达谱(GEP),得到包含16种细胞类型(每种1万个GEP)的平衡单细胞类型GEPs数据集S1。细胞比例可通过分段采样或随机采样生成细胞比例矩阵(P)。
- 批量肿瘤合成:以癌症基因组图谱(TCGA)包含19种癌症类型的数据集作为参考GEPs。结合S1和细胞比例矩阵(P)生成批量GEPs(D2/D0),再经过GEP过滤(与TCGA对比)和基因过滤(特征分数与细胞比例对比)两个过滤步骤,得到过滤后的批量GEPs(D1),作为训练集。
三、数据、材料和软件的可用性
软件实现与获取
DeSide使用Python和TensorFlow库构建深度神经网络模型,其软件包可在GitHub(https://github.com/OnlyBelter/DeSide )获取。
详细使用说明和文档可访问https://deside.readthedocs.io/ ,文档涵盖了DeSide的使用方法、研究中所用数据集信息以及主要函数参数。
代码与数据存储
用于重现研究结果和图表的源代码存于Zenodo存储库(https://zenodo.org/doi/10.5281/zenodo.7996661 ),预处理后的数据集也可在此获取。
公开数据集来源
- 批量RNA测序数据:GEO184398-Live中的数据从https://ftp.ncbi.nlm.nih.gov/geo/series/GSE184nnn/GSE184398/suppl/GSE184398_pancan_all_pc_genes_Live_TPM_Aug_3_20.tsv.gz下载;TCGA项目中19种肿瘤类型的批量RNA测序数据从GDC数据门户(https://portal.gdc.cancer.gov/ )以读取计数(htseq.counts)形式下载。
- 其他数据:图4A - C所用数据来自https://science.bostongene.com/kassandra/downloads;图4D中的原型流动群体值(即流动分数)从https://datalibrary.ucsf.edu/node/121/下载;包含基因长度信息的注释文件“gencode.gene.info.v22.tsv”从https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files下载。
单细胞RNA测序数据集
- 研究收集的部分scRNA - seq数据集(LUAD、GBM、PRAD)列于补充信息附录的数据集S6;
- OV - ascites从https://figshare.com/s/711d3fb2bd3288c8483a下载;
- HNSCC从3CA数据集(https://www.weizmann.ac.il/sites/3CA/head-and-neck )下载。
图表相关数据和代码
可在https://github.com/OnlyBelter/DeSide_mini_example/tree/main/plot_fig找到。
此外,研究还使用了先前已发表的数据,包括GEO184398-Live批量RNA测序数据、其他算法在3个批量RNA测序数据集上预测的细胞比例、TCGA中19种肿瘤类型的批量RNA测序数据、TCGA批量RNA测序数据的CPE值以及多个单细胞RNA测序数据。
四、项目演示
4-1:克隆仓库
# 使用HTTPS方式(推荐新手)
git clone https://github.com/OnlyBelter/DeSide_mini_example.git
# 进入项目目录
cd DeSide_mini_example
4-2:配置环境
# 创建conda环境(需提前安装Anaconda/Miniconda)
conda create -n deside python=3.8
conda activate deside
# 安装依赖包
pip install jupyterlab deside
4-3:下载数据文件
建议提前下载大文件(使用下载工具或浏览器直接下载):
预训练模型(示例1需要)
- 文件:model_DeSide.h5 (100MB)
- 下载地址:https://doi.org/10.6084/m9.figshare.25117862.v1
- 存放位置:创建目录并放入
mkdir -p DeSide_model # 将下载的文件移动到此目录
数据集文件(示例2和3需要)
-
示例2数据集 (2.2GB)
- 文件名:simu_bulk_exp_Mixed_N100K_D1.h5ad
- 下载地址:https://doi.org/10.6084/m9.figshare.23047391.v2
- 存放位置:
mkdir -p datasets/simulated_bulk_cell_dataset/D1 # 文件放入上述目录
-
示例3数据集 (7GB)
- 文件名:simu_bulk_exp_SCT_N10K_S1_16sct.h5ad
- 下载地址:https://doi.org/10.6084/m9.figshare.23043560.v2
- 存放位置:
mkdir -p datasets # 文件直接放入datasets目录
-
TCGA合并数据 (300MB)
- 文件名:merged_tpm.csv
- 下载地址:https://doi.org/10.6084/m9.figshare.23047547.v2
- 存放位置:
mkdir -p datasets/TCGA/tpm # 文件放入此目录
4-4:启动Jupyter Lab
conda activate deside
jupyter lab
浏览器会自动打开Jupyter界面,选择对应的示例笔记本
4-5:示例运行指南
示例1:使用预训练模型预测
- 打开
E1 - Using pre-trained model.ipynb - 检查内核是否选择
deside - 按顺序执行单元格:
- 加载模型:
model = load_model('DeSide_model/model_DeSide.h5') - 准备输入数据(需按格式要求)
- 运行预测:
predictions = model.predict(X_test)
- 加载模型:
- 结果保存在
results/example1/
示例2:从头训练模型
- 打开
E2 - Training a model from scratch.ipynb - 确保数据集路径正确:
data_path = "datasets/simulated_bulk_cell_dataset/D1/simu_bulk_exp_Mixed_N100K_D1.h5ad" - 重点步骤:
- 数据预处理
- 模型配置
- 训练过程监控(loss曲线)
- 训练完成后模型保存在
results/example2/
示例3:合成肿瘤样本
- 打开
E3 - Synthesizing bulk tumors.ipynb - 检查数据路径:
sct_gep_path = "datasets/simu_bulk_exp_SCT_N10K_S1_16sct.h5ad" tcga_path = "datasets/TCGA/tpm/merged_tpm.csv" - 关键步骤:
- 单细胞GEP筛选
- 参考TCGA数据过滤
- 合成bulk样本
- 结果输出到
results/example3/
4-6:常见问题解决
-
文件路径错误:
- 检查文件是否放在正确目录
- 使用绝对路径或调整相对路径
-
依赖包缺失:
# 在conda环境中重新安装 pip install -r requirements.txt -
内存不足:
- 关闭其他程序
- 使用云服务器(推荐16GB+内存)
-
下载中断:
- 使用wget续传:
wget -c [下载链接]
- 使用wget续传:
建议首次运行时先尝试示例1,熟悉流程后再运行需要大量资源的示例2和3。
科研合作意向统计
为了更好的利用小罗搭建的交流平台,我决定发放一个长期有效的问卷,征集大家在科研方面的任何需求,并且定期整理汇总,方便大家课题合作,招收学生,联系导师……
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









