







小罗碎碎念
这篇文章聚焦空间蛋白质组学领域,提出了首个基础模型KRONOS,旨在解决传统分析方法在多标记协同表达和复杂空间特征捕捉上的不足。
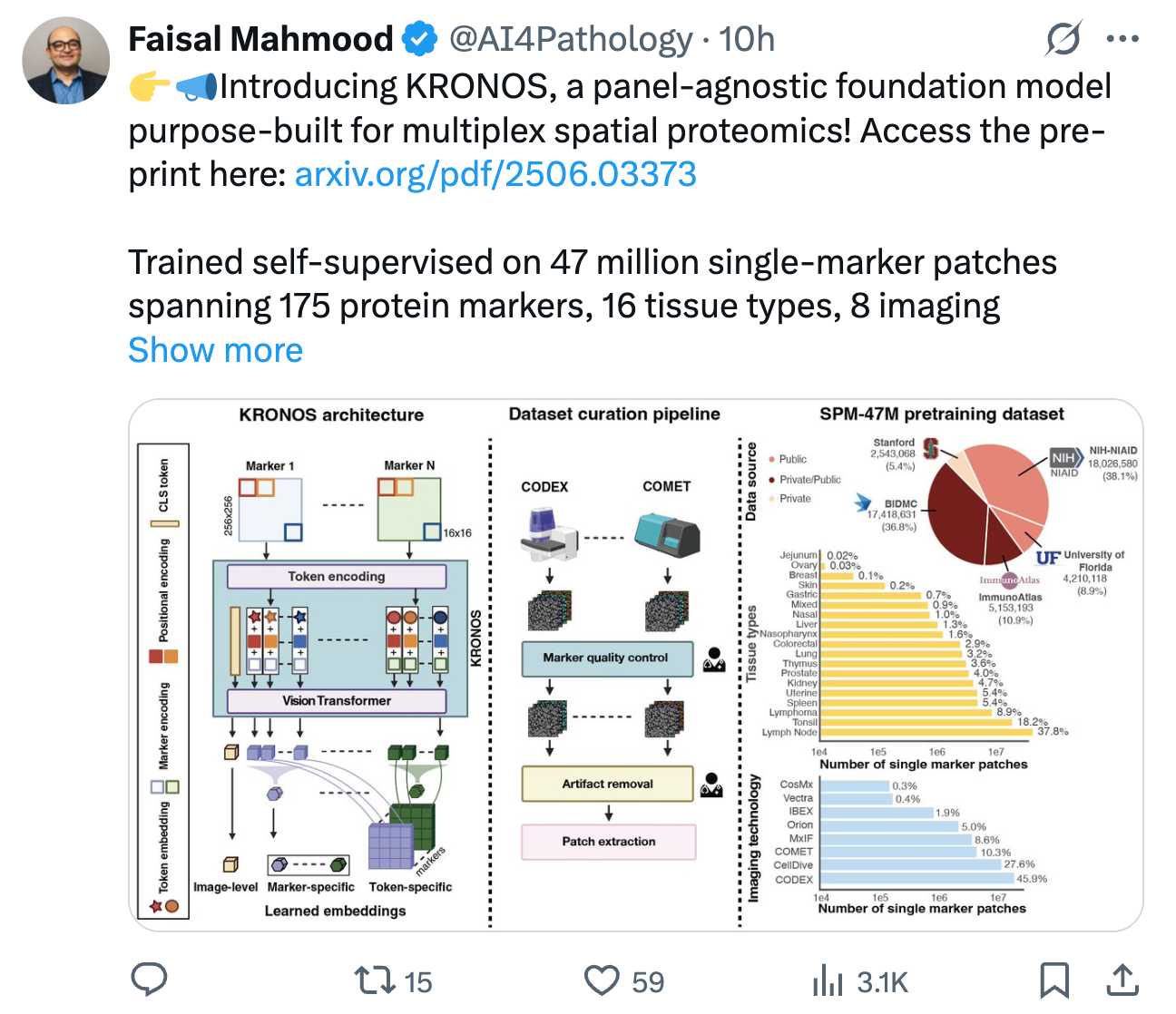
研究基于包含8种荧光成像平台、16种组织类型和175种蛋白质标记的4700万图像补丁数据集SPM-47M,采用Vision Transformer架构结合DINO-v2自监督学习策略,通过标记编码、位置编码和共享卷积嵌入层设计,使模型能够适应多通道、高维度数据,支持任意数量标记的动态输入,突破了传统模型对固定通道数的限制。
KRONOS在多项关键任务中展现出优异性能。在细胞表型分析中,其在经典霍奇金淋巴瘤和弥漫大B细胞淋巴瘤数据集上的平衡准确率显著优于通用模型和领域模型,且具备强大的少样本学习能力,仅用100个样本/类时仍能保持较高精度。
在组织微环境分析方面,KRONOS在区域分类、伪影检测和无监督聚类等任务中表现出色,例如在前列腺癌肿瘤与非肿瘤区域分类中AUC达0.91,伪影检测精确率达0.98。
此外,模型还能通过多实例学习实现患者治疗响应预测,并支持跨数据集的空间模式反向检索,推动跨研究的空间特征比较。
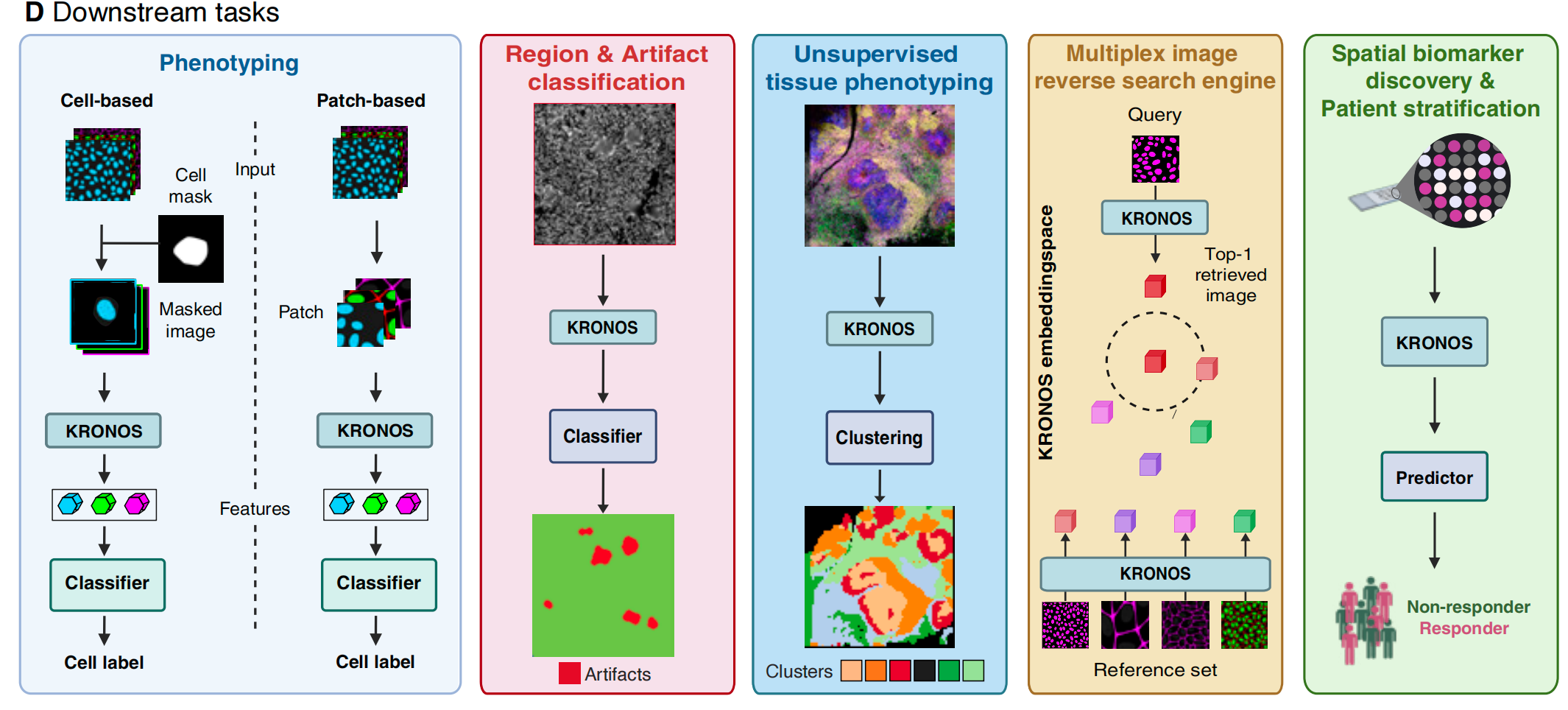
作为首个面向空间蛋白质组学的基础模型,KRONOS通过创新的无分割补丁分析范式降低了对细胞分割的依赖,提升了分析效率,且具备跨平台泛化能力,在8种成像技术和16种组织类型中表现一致。
模型代码与权重已公开,配套教程助力复现,为领域研究提供了可扩展的标准化分析框架。
尽管当前聚焦荧光成像,但其在数据高效性和多任务处理上的优势,为整合多组学数据、构建动态组织模型奠定了基础,有望加速医学AI在生物标志物发现和临床转化中的应用。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量67,000+,交流群总成员1500+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
加入我们
我们是一支以国内外硕博为主的团队,覆盖医学AI主流研究领域。
我们团队将持续招收有志于医学AI研究的青年学者,如需了解更多信息,欢迎扫码前往小罗的个人主页!
(建议在电脑端访问)
一、文献概述

1-1:研究背景与目标
空间蛋白质组学通过单细胞分辨率成像解析组织内蛋白质空间分布,对肿瘤微环境分析、治疗响应预测等具有重要意义。
然而,传统分析依赖细胞分割和单一标记阈值,难以捕捉多标记协同表达及复杂空间特征。
本文提出首个空间蛋白质组学基础模型KRONOS,通过大规模自监督学习突破传统方法的局限性,实现细胞表型分析、区域分类、患者分层等多任务的高效建模。
1-2:KRONOS模型构建
-
训练数据
- 数据集SPM-47M包含来自8种荧光成像平台、16种组织类型、175种蛋白质标记的4700万图像补丁,覆盖公共与内部队列,涵盖正常与恶性组织。
- 数据预处理包括人工质量控制(去除染色噪声、组织折叠等)、组织掩膜提取有效区域,确保多源数据的一致性。
-
架构设计
- 基于Vision Transformer (ViT)架构,采用DINO-v2自监督学习策略,结合标记编码(sinusoidal编码区分不同蛋白质标记)、位置编码和共享卷积嵌入层,适应多通道、高维度的空间蛋白质组学数据。
- 创新点:支持任意数量标记的动态输入,通过标记无关的共享权重和可扩展的标记编码,解决传统模型对固定通道数的依赖。
1-3:核心功能与性能验证
-
细胞表型分析
- 在经典霍奇金淋巴瘤(cHL)和弥漫大B细胞淋巴瘤(DLBCL)数据集上,KRONOS的细胞表型分类平衡准确率显著优于通用模型(如DINO-v2、UNI)和领域模型(CA-MAE)。例如,在DLBCL-2中,KRONOS平衡准确率达0.7969,远超DINO-v2的0.2980。
- 少样本学习能力:仅用100个样本/类时,KRONOS在cHL中平衡准确率达0.6735,而DINO-v2仅为0.3217,显示其高效的数据利用能力。
-
无分割表型分析
- 提出补丁级分析范式,无需细胞分割即可通过32×32像素补丁预测主要细胞表型,平衡准确率比传统标记强度均值方法高10%-20%,尤其在标记噪声较高的场景下优势显著。
-
组织微环境分析
- 区域分类:在前列腺癌数据中,KRONOS通过补丁特征区分肿瘤与非肿瘤区域,AUC达0.91,显著高于标记强度基线(0.68)。
- 伪影检测:利用DAPI通道识别组织折叠、信号饱和等伪影,精确率达0.98,远胜传统方法(0.74)。
- 无监督聚类:在结直肠癌中,KRONOS通过补丁特征聚类成功区分克罗恩样反应(CLR)与弥漫性炎症浸润(DII)微环境,聚类结果与临床表型高度吻合(ARI值最高)。
-
患者分层与图像检索
- 在肾细胞癌(ccRCC)和皮肤T细胞淋巴瘤(CTCL)中,KRONOS通过多实例学习实现治疗响应预测,AUC最高达0.7895(ccRCC),优于通用模型。
- 开发空间模式反向搜索引擎,支持跨数据集的细胞/补丁检索,例如在DLBCL中检索EBV阳性肿瘤微环境,Top-1准确率达0.8233,推动跨研究的空间特征比较。
1-4:创新与意义
-
方法创新
- 首次将基础模型引入空间蛋白质组学,解决多通道、异质性数据的表征学习问题。
- 提出无分割补丁分析范式,降低对高质量细胞分割的依赖,提升分析效率。
-
应用价值
- 数据高效性:在标记成本高昂的空间 proteomics中,KRONOS通过少样本学习和无监督聚类减少人工标注需求。
- 跨平台泛化:在8种成像技术、16种组织类型中表现一致,支持多中心数据整合。
- 开源与工具化:模型代码与权重公开(https://github.com/mahmoodlab/KRONOS),配套教程助力复现,推动领域标准化。
1-5:局限与展望
当前模型聚焦荧光成像,未来计划扩展至离子成像(如IMC、MIBI),并整合转录组等多组学数据,构建动态组织模型。
KRONOS为空间蛋白质组学提供了可扩展的分析框架,有望加速生物标志物发现与临床转化。
二、KRONOS概述
什么是KRONOS?
KRONOS是一种与标记组合无关的空间蛋白质组学基础模型,支持对不同实验设置下的多重成像数据进行分析。
该系统在跨越175种蛋白质标记、16种组织类型、8种成像平台和5家机构的4700万个单标记图像补丁上进行自监督训练。
关键特性
| 特性 | 描述 |
|---|---|
| 架构 | 基于视觉Transformer,采用共享通道主干和正弦标记身份嵌入 |
| 训练规模 | 覆盖175种蛋白质标记的4700万个补丁 |
| 标记组合兼容性 | 原生支持不同机构的可变标记组合 |
| 输出类型 | 补丁嵌入、标记嵌入和令牌嵌入 |
| 部署方式 | 通过HuggingFace Hub提供,支持create_model_from_pretrained接口加载 |
2-1:系统架构概述
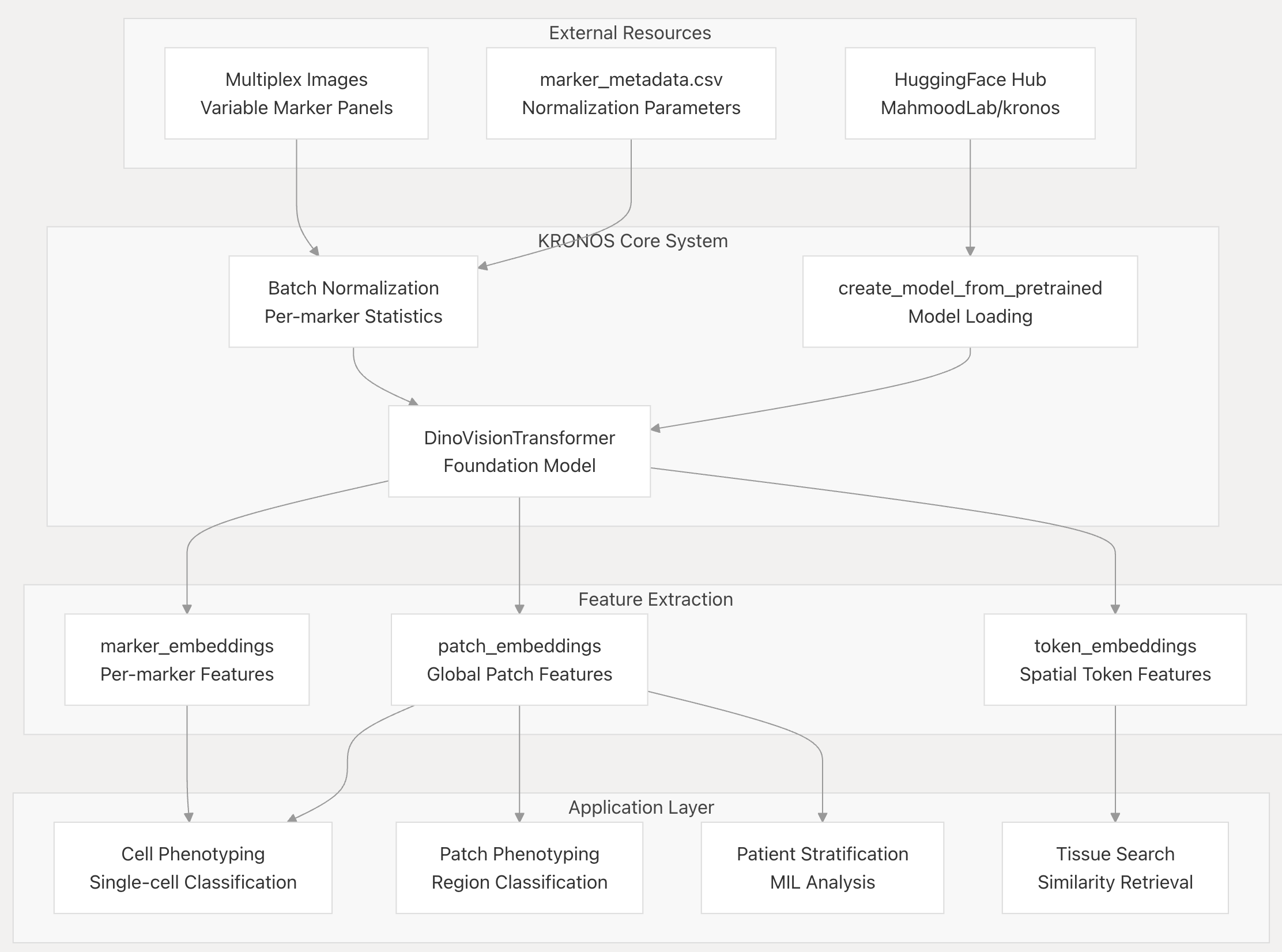
- External Resources(外部资源):Multiplex Images(多重图像,含可变标记组合 )、marker_metadata.csv(标记元数据文件,含归一化参数 )、HuggingFace Hub(模型仓库,存储MahmoodLab/kronos )
- KRONOS Core System(KRONOS核心系统):Batch Normalization(批归一化,基于标记统计 )、create_model_from_pretrained(模型加载函数 )、DinoVisionTransformer(基础模型 )
- Feature Extraction(特征提取):marker_embeddings(标记级特征 )、patch_embeddings(全局补丁特征 )、token_embeddings(空间令牌特征 )
- Application Layer(应用层):Cell Phenotyping(单细胞分类 )、Patch Phenotyping(区域分类 )、Patient Stratification(患者分层,基于多实例学习分析 )、Tissue Search(组织检索,基于相似性 ) ,呈现了从数据输入到特征提取、再到应用的完整流程。
2-2:核心组件和代码实体
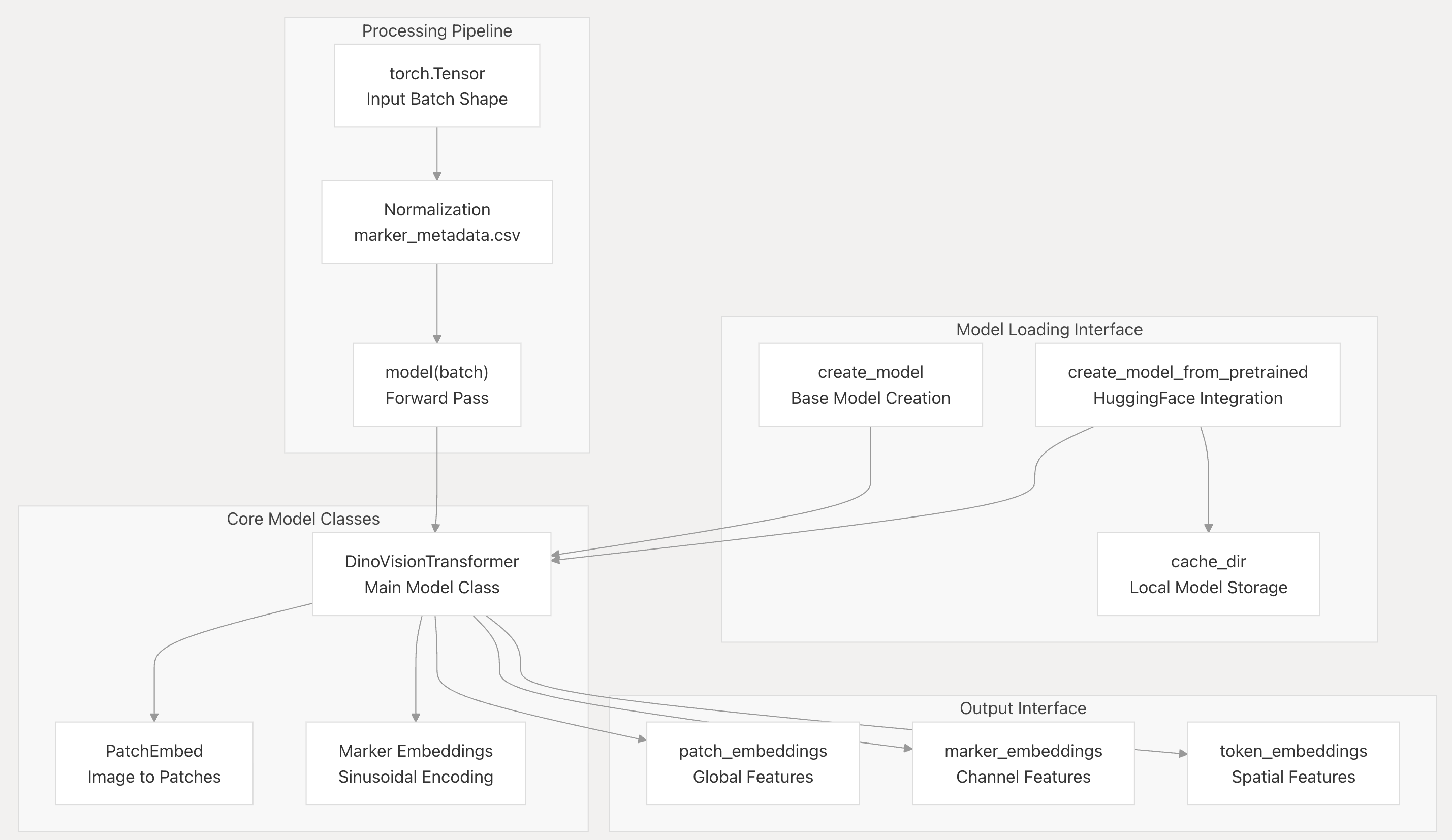
- Processing Pipeline(处理流程):输入为torch.Tensor(Input Batch Shape ),经Normalization(基于marker_metadata.csv )、model(batch) Forward Pass(前向传播 )步骤
- Model Loading Interface(模型加载接口):含create_model(基础模型创建 )、create_model_from_pretrained(HuggingFace集成 ),关联cache_dir(本地模型存储 )
- Core Model Classes(核心模型类):主类DinoVisionTransformer,关联PatchEmbed(图像转补丁 )、Marker Embeddings(正弦编码 )
- Output Interface(输出接口):输出patch_embeddings(全局特征 )、marker_embeddings(通道特征 )、token_embeddings(空间特征 ) ,呈现模型从输入处理、加载到特征输出的完整流程 。
2-3:关键功能
标记组合无关处理
KRONOS通过共享通道主干架构结合正弦标记身份嵌入处理具有可变标记组合的多重图像。
这一设计使其无需重新训练即可处理不同机构和成像平台的数据。
多尺度特征提取
模型生成三种类型的嵌入以支持全面分析:
| 嵌入类型 | 描述 | 典型用例 |
|---|---|---|
patch_embeddings | 全局补丁级特征 | 患者分层、组织分类 |
marker_embeddings | 单标记通道特征 | 细胞表型分析、标记分析 |
token_embeddings | 空间令牌特征 | 组织检索、空间分析 |
可扩展处理
KRONOS支持可配置参数的批处理:
batch_size = 2
marker_count = 10 # 随数据集可变
patch_size = 224 # 固定输入尺寸
2-4:应用领域
KRONOS支持六种主要应用工作流程:
监督应用
- 细胞表型分析:使用
marker_embeddings进行单细胞分类 - 补丁表型分析:无需细胞分割的组织区域分类
- 患者分层:基于多实例学习的患者水平预测
无监督应用
- 组织表型分析:基于细胞组成的组织区域聚类
- 区域与伪影检测:质量控制和技术噪声识别
- 组织检索:形态和免疫特征的相似性检索
每种应用均利用核心模型生成的三种嵌入类型的不同组合。
2-5:模型访问和要求
硬件要求
- 消费级GPU(如NVIDIA 3090)
- 兼容标准PyTorch安装
软件要求
- Ubuntu 22.04(经测试)
- Python 3.10
- PyTorch生态系统
模型访问
预训练模型通过HuggingFace Hub发布(MahmoodLab/kronos),需申请访问权限。create_model_from_pretrained函数支持自动下载和缓存。
三、KRONOS 模型使用指南
3-1:系统要求与安装
-
硬件要求
- 支持 CUDA 的 NVIDIA GPU(推荐 RTX 3090 或更高)
- 至少 24GB GPU 内存(用于处理大尺寸图像)
-
软件环境
# 1. 创建虚拟环境(任选一种方式) # Conda方式 conda create -n kronos python=3.10 conda activate kronos # venv方式(Linux/macOS) python3 -m venv kronos source kronos/bin/activate # 2. 安装 KRONOS git clone https://github.com/mahmoodlab/KRONOS.git cd KRONOS pip install -e .[tutorials] # 安装核心库+教程依赖
3-2:模型获取与初始化
-
申请模型权限
- 访问 HuggingFace 页面
- 点击 “Request Access” 提交申请
- 获得授权后下载模型文件(约 5GB)
-
加载预训练模型
from kronos import create_model_from_pretrained # 加载模型 (首次运行会自动下载) model, precision, embed_dim = create_model_from_pretrained( checkpoint_path="hf_hub:MahmoodLab/kronos", cache_dir="./kronos_weights" # 模型保存路径 ) # 检查设备兼容性 device = "cuda" if torch.cuda.is_available() else "cpu" model = model.to(device) print(f"Loaded {precision} model | Embedding dim: {embed_dim}")
3-3:数据处理流程
-
数据规范要求
- 输入格式:
[batch_size, num_markers, height, width] - 建议 patch 尺寸:224×224 像素
- 标记通道顺序:按标记 ID 排序
- 输入格式:
-
数据标准化处理
import torch import numpy as np # 假设已加载图像数据 (10个标记) raw_data = np.load("multiplex_image.npy") # 形状: [10, 224, 224] # 转换为PyTorch张量 batch = torch.tensor(raw_data).unsqueeze(0).to(device) # 添加batch维度 # 从元数据获取标准化参数(示例) marker_means = torch.rand(10).to(device) # 替换为实际均值 marker_stds = torch.rand(10).to(device) # 替换为实际标准差 # 执行通道标准化 normalized_batch = (batch - marker_means[None, :, None, None]) / marker_stds[None, :, None, None]
3-4:特征提取
# 启用推理模式 (节省内存)
with torch.no_grad():
patch_emb, marker_emb, token_emb = model(normalized_batch)
# 检查输出维度
print("Patch embeddings:", patch_emb.shape) # [batch, patch_emb_dim]
print("Marker embeddings:", marker_emb.shape) # [markers, marker_emb_dim]
print("Token embeddings:", token_emb.shape) # [batch, tokens, token_emb_dim]
# 保存特征供下游任务使用
np.save("patch_embeddings.npy", patch_emb.cpu().numpy())
3-5:核心应用场景
-
细胞分型(Cell Phenotyping)
- 所需文件:
tutorials/2 - Cell-phenotyping.ipynb - 关键步骤:
- 提取细胞级嵌入特征
- 训练浅层分类器(SVM/Random Forest)
- 可视化细胞类型分布
- 所需文件:
-
组织搜索(Tissue Search)
# 余弦相似度计算 from sklearn.metrics.pairwise import cosine_similarity query_emb = patch_emb[0].cpu().numpy() # 查询样本 db_embeddings = np.load("database_embs.npy") # 数据库 # 计算相似度 similarities = cosine_similarity([query_emb], db_embeddings)[0] top_matches = np.argsort(similarities)[-5:][::-1] # 可视化前5个匹配结果 show_match_results(top_matches) -
无监督组织分型
from sklearn.cluster import KMeans # 使用所有patch嵌入 all_embeddings = np.vstack([patch_emb1, patch_emb2, ...]) # 聚类分析 kmeans = KMeans(n_clusters=8) cluster_labels = kmeans.fit_predict(all_embeddings) # 可视化聚类结果 plot_tissue_clusters(whole_slide_image, cluster_labels)
3-6:典型问题解决方案
-
CUDA 内存不足
# 解决方案1: 减小batch size model.inference(batch_size=2) # 解决方案2: 混合精度训练 from torch.cuda.amp import autocast with autocast(): embeddings = model(batch) -
标记通道不匹配
- 创建映射字典重新排序通道
- 缺失标记用零填充
# 示例通道映射 channel_map = {'CD3': 0, 'CD20': 1, ...} ordered_batch = torch.zeros_like(batch) for i, marker in enumerate(marker_names): ordered_batch[:, channel_map[marker]] = batch[:, i]
3-7:迁移学习配置
# 冻结基础层
for param in model.encoder.parameters():
param.requires_grad = False
# 替换分类头
model.classifier = torch.nn.Linear(embed_dim, num_classes)
# 微调最后一层
optimizer = torch.optim.Adam(model.classifier.parameters(), lr=1e-3)
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎扫码前往小罗的个人主页!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









