







题目:scRMD: imputation for single cell RNA-seq data via robust matrix decomposition
出处:bioinformatics, doi: 10.1093/bioinformatics/btaa139
摘要:单细胞rna测序技术使人们能够在单细胞级别的分辨率下进行转录组分析,然而由于其常常难以捕捉到表达的基因,因此会导致显著的dropout(姑且译为缺失值)问题,从而影响下游分析,例如使差异表达分析的统计功效降低以及模糊了细胞之间、基因之间的关系。本文将缺失值的插值问题建模为矩阵分解问题,提出scRMD算法,实验证明该算法能够准确地还原缺失值并有助于下游分析,如差异表达分析和细胞聚类。
主要思想:令基因表达矩阵的观测值为p行n列的矩阵Y,其中p是基因数,n是细胞数,表达矩阵的真实值为X,dropout矩阵为S,即,若Xij处出现了dropout现象则Sij=Xij,否则为0,E是期望值为0的随机误差矩阵,故有
,
这里的一个关键点是,表达矩阵X中虽然可能包含许多细胞,但可以分为少数几个类,基因在不同类的细胞中的期望表达水平不同,故有
其中li,c(j)是第j个细胞所属的细胞类型中基因i的期望表达水平,fi,j是随机误差,表示为矩阵形式有
。
这里可以注意到由于细胞的种类数要远少于细胞个数,因此L是低秩矩阵,从而可以通过最小化
来估计低秩的细胞类型矩阵L和dropout矩阵S,其中分别是Frobenious范数,核范数和L1范数。本文使用基于随机矩阵的技术来选择参数
和
。
主要实验结果:
(1)模拟数据的差异表达分析与聚类:
使用不同的正态分布生成代表不同细胞类型的模拟数据,并加入dropout,对生成的模拟数据进行插值,结果如图:

(2)真实数据差异表达分析:
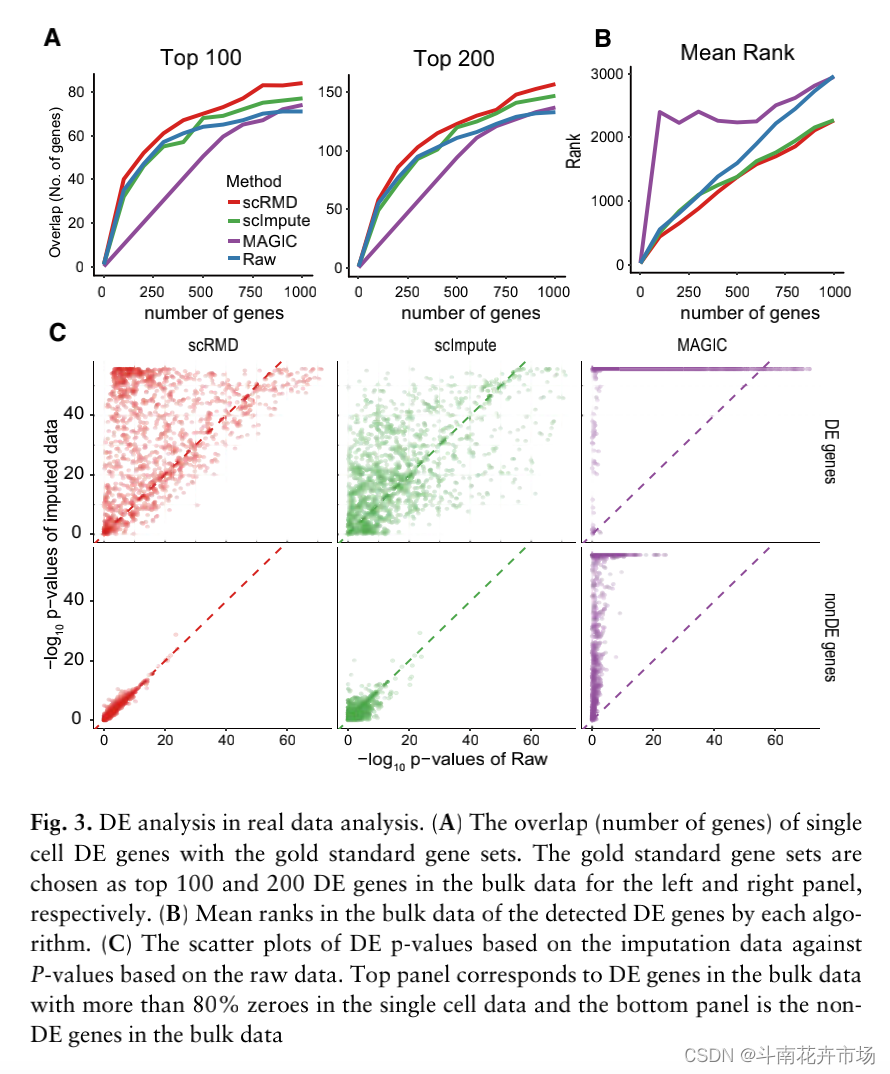
如上图,以bulk rna-seq的数据作为金标准,对单细胞数据进行插值后进行差异表达分析,提取p值最小,即最显著的差异表达基因与金标准进行比较,比较两者得到的差异表达基因。
(3)ERCC基因相关系数:
ERCC基因是人为加入的一些已知浓度的基因,用于作为参照物校正其它基因的表达水平,因此可以比较插值前后ERCC基因观测值与真实值的相关系数来衡量插值效果,在5种不同测序技术的数据中的结果如下图所示:

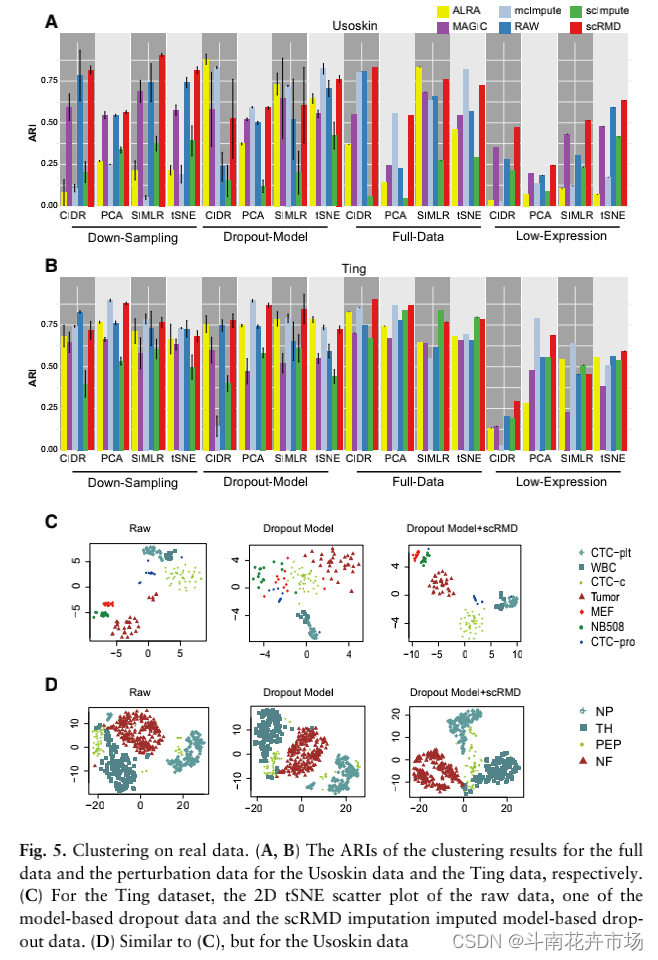
(4)真实数据中的聚类结果:
在两个不同数据中进行插值,并比插值前后的聚类结果,使用调整兰德指数作为指标,结果如下所示:
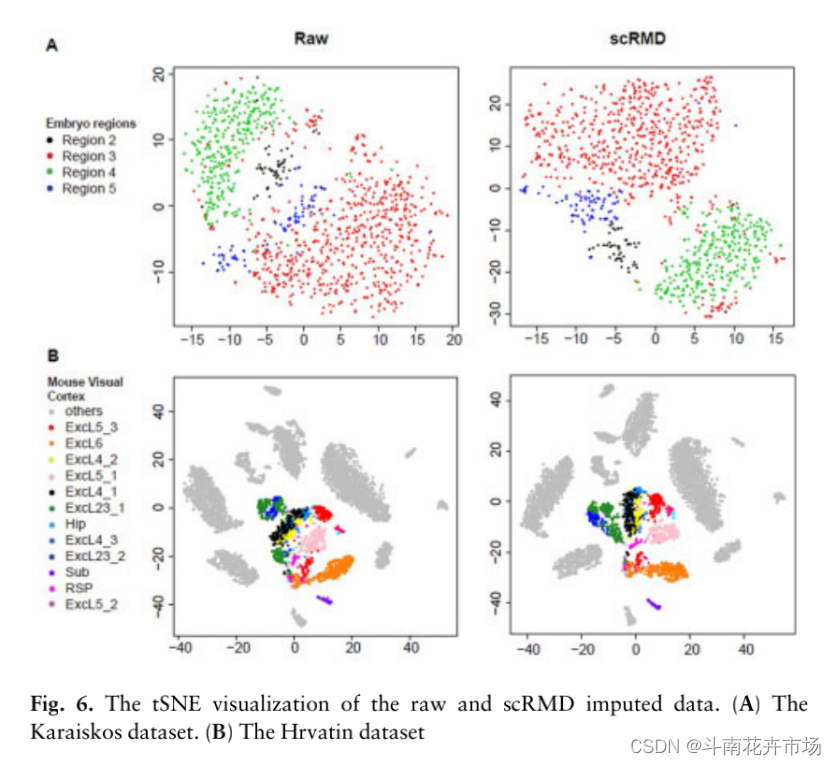
(5)超高通量数据插值结果可视化:
超高通量数据通常使用唯一分子标识符(UMI)以去除扩增偏好造成的表达水平失真,采用这种技术的数据通常存在更多的零值。本文考虑三个超高通量数据,正文中给出其中两个的tSNE可视化结果,如下图所示:


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









