






 本文介绍如何使用MATLAB批量读取同一文件夹内以.counts为结尾的TXT文件,并将它们合并成一个包含基因ID和表达值的数据框。通过dir查找文件名,构建文件路径,然后用lapply和read.table读取文件,最后利用do.call(cbind, a)进行列合并。"
112278597,2820065,Flink SQL中DataStream与Table的全局窗口Join操作解析,"['flinksql', 'join', 'table']
本文介绍如何使用MATLAB批量读取同一文件夹内以.counts为结尾的TXT文件,并将它们合并成一个包含基因ID和表达值的数据框。通过dir查找文件名,构建文件路径,然后用lapply和read.table读取文件,最后利用do.call(cbind, a)进行列合并。"
112278597,2820065,Flink SQL中DataStream与Table的全局窗口Join操作解析,"['flinksql', 'join', 'table']
先描述一下情况,我有一个文件夹叫“原始1”
打开看一看,里面有大坏蛋 和小可爱文件
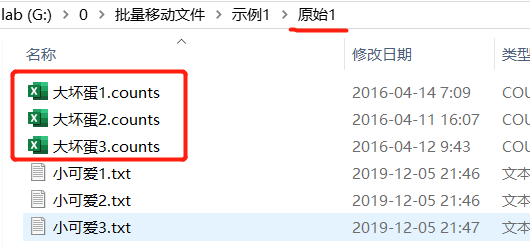
3个大坏蛋文件,打开格式如下, 第一列基因ID一模一样,区别是第二列不同。
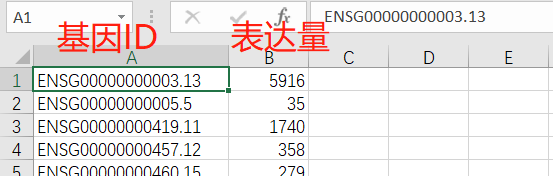
需求:
我们想把3个大坏蛋读取,合并成一个文件:第一列是基因ID,后面列都是表达值
批量读取文件三部曲:
1.找到文件名
2.构建文件的路径
3.lapply配合read.table读取
1. 找到文件名
dir查看‘原始1’里东西
p1'原始1',pattern = '*.counts')大坏蛋名字都是.counts结尾,所以pattern参数指定读取counts结尾数据

2. 构建文件路径
构建文件目录
p2'原始1/',p1)
3. lapply 配合 read.table读取
arow.names=1 意思是 原来的第一列,读取当做行名
返回的a是一个list, 这个list相当于一个柜子,每个抽屉存放着读取的3个文件
list 转 data.frame
a1<-data.frame(do.call(cbind,a))do.call(cbind, a) 就是把 a这个list, 里面文件内容 按cbind列合并,返回data.table
所以进一步data.frame转换成数据框
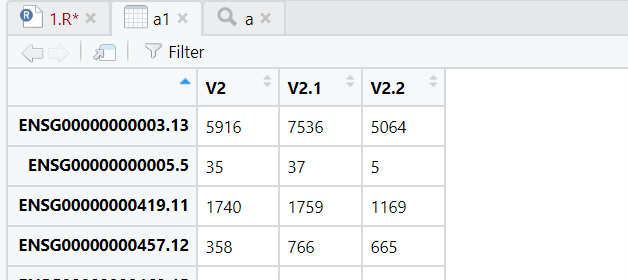
大坏蛋读取、合并就此解决



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









