






高岭石是矿山尾矿废水中的重要粘土成分,高岭石经常被用来模拟含高悬浮固体的尾矿废水,高比表面积,强电负性和水合特性的高岭土细颗粒(小于2μm)是形成稳定悬浮液的重要原因,它对尾矿的沉降和脱水产生很大的影响。如果对尾矿中的粘土矿物进行不当的处理和处置,将会造成水资源的严重浪费,环境二次污染以及整个工业生产过程的损害,例如水质下降和浮选产品精度下降。丙烯酰胺(PAM)广泛用于各种废水处理行业,其功能是将废水转化为有用的废水,然后再返回水循环。据报道,已经对各种类型的PAM和高岭石吸附絮凝进行了广泛的研究。PAM和高岭石之间的相互作用是氢键,电荷中和,电桥和静电吸附,这得到了大多数研究人员的支持。阳离子聚丙烯酰胺(CPAM)似乎比其他用于处理高岭石悬浮固体的试剂更受青睐,可能发生在带有阳离子电荷的CPAM与带有负电荷的高岭石颗粒表面之间发生电荷中和但是,宏观沉降试验在解释吸附机理如何方面仍然遇到许多困难。
PAM是一种高分子量线性水溶性聚合物,通常通过将丙烯酰胺(AM)与有机单体共聚而制得。根据水溶液中PAM的离子特性,PAM有四种不同类型:非离子型,阴离子型,阳离子型和两性离子型,非离子聚丙烯酰胺(NPAM)能显著降低工业废水混凝过程中的污泥量,提高染料去除速度。阴离子聚丙烯酰胺(APAM)在去除浊度方面表现出优异的效率,可用于处理金属尾矿或含重金属的废水。 CPAM是去除循环水中高悬浮固体的最有效絮凝剂,它被广泛用于生物废水的预处理和活性污泥的脱水,除此之外,CPAM已被证明在处理含油废水方面令人满意。除废水处理外,PAM在造纸,二次采油,生物医学和土壤改良等方面也具有广阔的应用前景。
最近,分子模拟已广泛用于在分子/原子水平上深入了解粘土矿物与有机物之间的相互作用。其中,过去曾使用分子动力学(MD)模拟来研究水溶液中的高岭石表面特征,从高岭石结构到H2O在高岭石表面上的吸附行为,再到吸附在高岭石表面上的小分子的电子结构性质,DFT已被广泛认可和使用。迄今为止,尽管研究人员已尝试合成不同类型的CPAM以处理高浓度废水。对于悬浮固体,如何正确选择CPAM聚合的单体尚不清楚,主要原因是CPAM的物理和化学细节以及高岭石的吸附作用尚未得到充分记录。
因此,安徽理工大学团队这项工作旨在详细了解CPAM结构单元如何作用于高岭土基体表面的吸附机理,这将对目标聚合单体的选择产生深远影响,并促进实验和理论研究相结合,以去除废水中的高悬浮固体。该研究结果以“Adsorption mechanism insights into CPAM structural units on kaolinite
surfaces: A DFT simulation”为题发表在«Applied Clay Science »上(见文后原文链接)。

【图文解析】
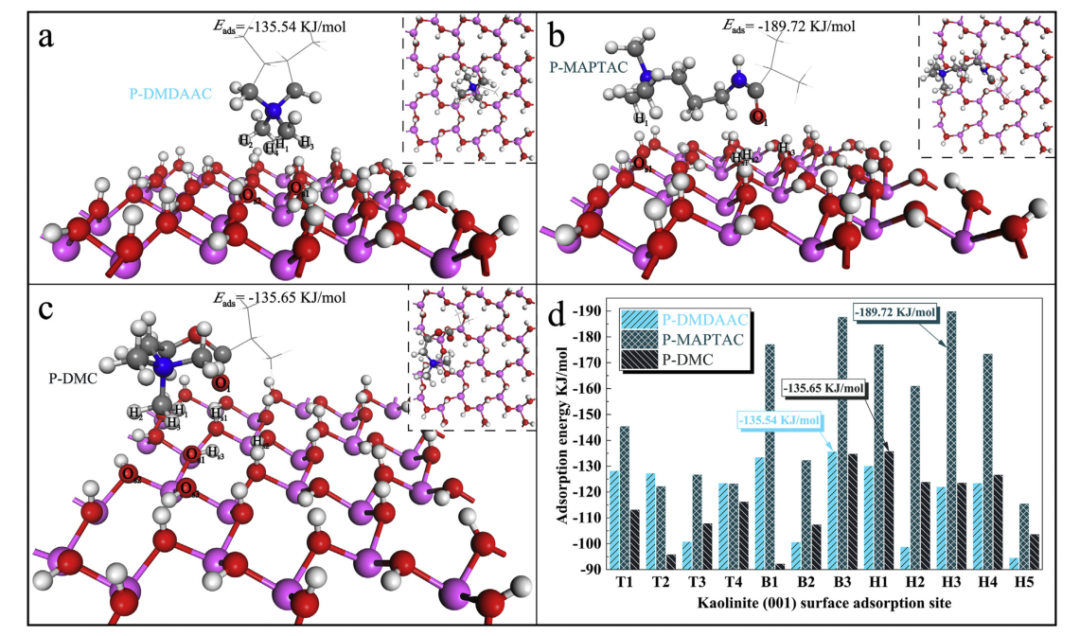
图1.高岭石(001)表面上CPAM阳离子结构单元的吸附能和最佳吸附构型。
a:P-DMDAAC /高岭石(001)表面,b:P-MAPTAC /高岭石(001)表面,c:P-DMC /高岭石(001)表面,d:CPAM结构单元在最佳姿势下的吸附能高岭石(001)表面。
解析
作者考虑了三种常见的CPAM:P(AM-DMDAAC),P(AM-MAPTAC)和P(AM-DMC)。关于CPAM结构单元在高岭石(001)表面吸附位点上最佳姿势的吸附能如图1d所示,如所观察到的,所有吸附模型的吸附能均为负,表明在CPAM结构单元吸附过程中发生了放热过程,众所周知,吸附能越低,吸附能力越强。重要的是要注意,CPAM作为聚合物,结构单元可能被高岭石表面上的任何潜在吸附位点吸附。我们发现P-MAPTAC在高岭石(001)表面的大多数吸附位点上显示出强相互作用。 P-DMDAAC在顶部位点和桥位点的结合亲和力高于P-DMC,但在空心位点相反。考虑到P-DMDAAC,P-MAPTAC和P-DMC在高岭石(001)表面上的平均吸附能分别为-118.04,-152.44和-114.96 KJ / mol。总体而言,这三个阳离子结构单元在高岭石(001)表面上的吸附能力的顺序为P-MAPTAC> P-DMDAAC> P-DMC。
图1a,b和c分别显示了三种CPAM阳离子结构单元在高岭石(001)表面上的最佳吸附构型。相应的吸附能分别为-135.54,-189.72和-135.65 KJ / mol。仅从吸附结构的几何形状观察。 P-DMDAAC和高岭石(001)表面的吸附系统最可能结合的原子是Os1,H1,H3和Os2,H2,H4。当P-MAPTAC吸附在高岭石(001)表面上时,高岭石(001)表面的原子(Hs1,Hs2,Hs3和Os1)与P-MAPTAC的O1和H1之间可能发生键相互作用。同样,P-DMC在高岭石(001)表面上可能的键合相互作用分别位于Hs1,Hs2,Hs3,O1和Os1,H1和Os2,H2和Os3,H3。

图2. CPAM阳离子结构单元在高岭石(001(-))表面上的吸附能和最佳吸附构型。
a:P-DMDAAC /高岭石(001(-))表面,b:P-MAPTAC /高岭石(001(-))表面,c:P-DMC /高岭石(001(-))表面,d:CPAM结构单元在最佳姿势下的吸附能高岭石(001(-))表面。
解析
图2d说明了CPAM结构单元在高岭石(001(-))表面上最佳姿势的吸附能。 P-DMDAAC,P-MAPTAC和P-DMC在高岭石(001(-))表面上的平均吸附能分别为-186.10,-226.34和-185.13 KJ / mol。因此,高岭石(001(-))表面上的吸附能力具有以下顺序:PMAPTAC> P-DMDAAC> P-DMC。不难得出这样的结论,即结构单元在高岭石(001(-))表面的吸附稳定性高于高岭石(001)表面的吸附稳定性。根据吸附能的值(图1d和2d),高岭石(001)和(001(-))表面上三个CPAM结构单元的平均吸附能分别为-147.21,-184.07和-145.03 KJ / mol 。因此,我们可以安全地得出结论,即P-MAPTAC与高岭石表面的最佳结合相互作用。 CPAM阳离子结构单元在高岭石(001(-))表面上的最佳吸附结构分别如图2a,b和c所示。三种吸附模型的共同点是,最佳吸附位为H3,相应的吸附能分别为-200.56,-238.25和-199.05 KJ / mol。我们还在图中标记了最可能具有键相互作用的原子。然而,仅键合位置和键合原子可以通过吸附系统的几何构型来分析,并且键合的性质需要进一步分析,尤其是参考键合性质。
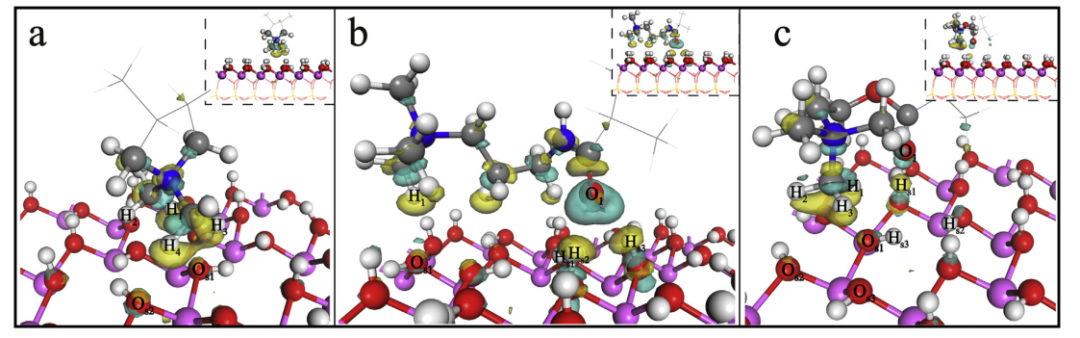
图3.高岭石(001)表面上CPAM结构单元的电子密度差。
a:P-DMDAAC /高岭石(001)表面; b:P-MAPTAC /高岭石(001)表面; c:P-DMC /高岭石(001)表面,其等值为0.01电子/Å3。
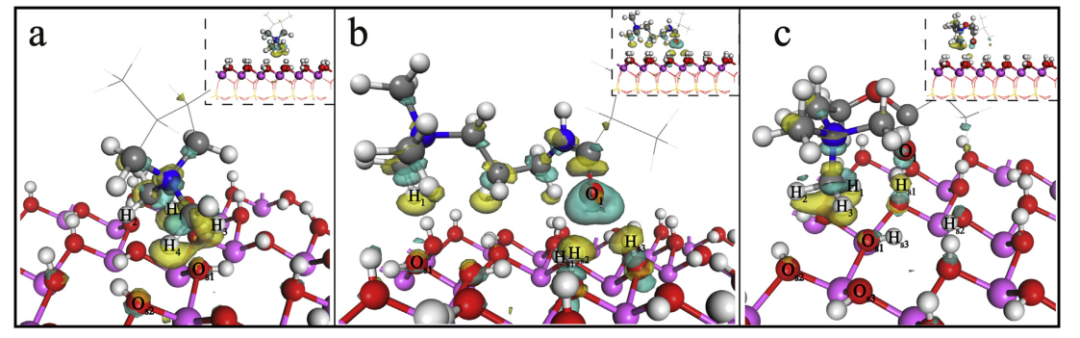
图4.高岭石(001-)表面上CPAM结构单元的电子密度差。
a:P-DMDAAC /高岭石(001(-))表面; b:P-MAPTAC /高岭石(001(-))表面; c:P-DMC /高岭石(001(-))表面,其等值为0.01电子/Å3。
解析
图3和图4示出了高岭石表面上的结构单元的电子密度差。青色区域表示电子积累,而黄色区域表示电子耗尽。P-DMDAAC /高岭石(001)表面的吸附系统显示H1和H4原子获得0.03和0.04|e|,Os1和Os2均获得0.04|e|。表面氢原子Hsn(n = 2和3)和Os1分别获得或损失0.01|e|;并且结构单元的O1损失0.03|e|和H1获得0.02|e| P-MAPTAC吸附在高岭石(001)表面上之后。参照P-DMC吸附,高岭石(001)表面原子Hs1和Os1分别获得0.01和0.02|e|。 P-DMC中的H1原子损失0.02|e| O1获得0.02|e|。高岭石(001)表面上CPAM结构单元的键原子的电子变化如图3所示。它们的共同特征是,当氢原子的电子耗尽时,氧中会发生电子积累H…O表示强结合亲和力。此外,结构单元中的氧(Om)和表面氢(Hsn)的电子累积或耗尽区大于结构单元中的氢(Hm)和表面氧(Osn)。我们认为氢键O-Hsn…Om比C-Hm…Osn强。总之,P-DMDAAC /高岭石(001)表面的吸附是由两个氢键驱动的,即C-H1…Os1和C-H4…Os2。当P-MAPTAC吸附在高岭石(001)表面上时,形成三个氢键(C-H1…Os1,O-Hs2…O1和O-Hs3…O1)。对于P-DMC吸附,氢键为O-Hs1…O1和C-H1…Os1。 Chen等认为,大量电子在被吸附物和吸附剂之间转移,吸附系统越稳定,静电吸引力越大(Chen et al.,2017)。从P-DMDAAC,P-MAPTAC和P-DMC到高岭石(001)表面的电荷转移量分别为0.56, 0.25和0.23|e|。这表明CPAM结构单元在高岭石(001)表面上的静电吸引顺序为P-DMDAAC> P-MAPTAC> PDMC。显然,静电吸引发生并主导了该过程。
基于吸附容量的分析,从P-DMDAAC,P-MAPTAC和P-DMC到高岭石(001(-))表面的电荷转移量分别为0.90、0.89和0.89|e|。我们观察到,当结构单元吸附在高岭石(001(-))的表面上时,除了(001(-))表面原子上电子的耗竭或积累,甚至影响高岭石的内部和表面(001)(图4)。电荷转移是造成这种现象的原因。三个CPAM结构单元主要是高岭石(001(-))表面上的静电吸引。吸附系统的稳定性高于高岭石(001(-))表面的稳定性。这些结果与吸附能分析一致。

图5. P-DMDAAC /高岭石(001)表面的键合原子的PDOS。费米能级用黑色虚线表示。a:H1和Os1,b:H4和Os2,拖影值为0.01。
解析
进行了键合原子的PDOS投影,以进一步探索高岭石表面上结构单元吸附的键合性质。PDMDAAC /高岭石(001)表面中C-Hm…Osn(m = 1,4和n = 1,2)氢键的性质如图5所示。吸附之前这表明吸附后键合原子更稳定。在价带处,在Hm-1s和Osn-1s与2p轨道之间观察到轨道重叠,范围约为-10 eV至-7 eV和0 eV至3 eV。 Hm和Osn曲线在占用的PDOS配置文件中具有类似的趋势。这些结果证实在P-AM /高岭石(001)表面上形成了C-H1…Os1和C-H4…Os2的氢键。
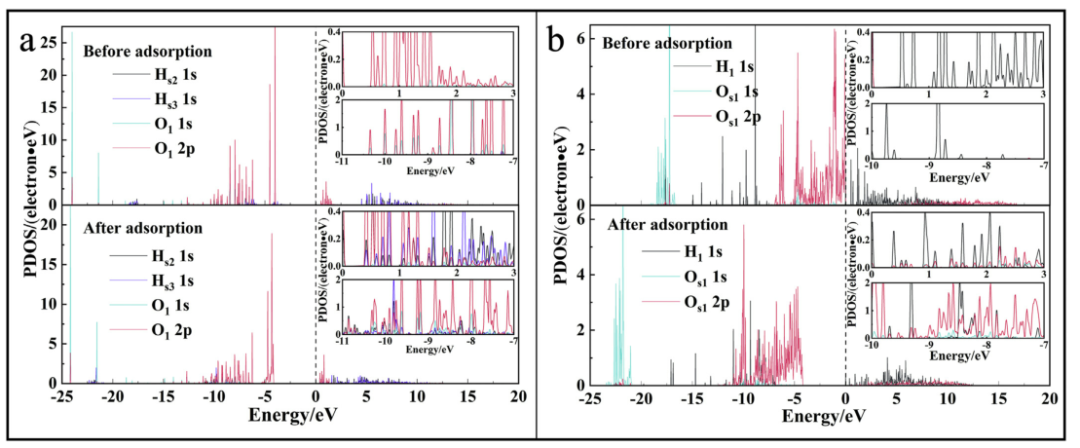
图6. P-MAPTAC /高岭石(001)表面的键合原子的PDOS。费米能级用黑色虚线表示。键原子:Hs1,Hs2,Hs3和O1,涂片值为0.01。
解析
P-MAPTAC和高岭石(001)表面的氢键的PDOS如图6所示。键原子的PDOS峰下降,而价带变宽,离域增强。 O1和Hsn(n = 2和3)的键在-11 eV至-7 eV和0 eV到3 eV的范围内,吸附前Hsn的轨道很少出现,但键原子在吸附后形成相同的共振。在氢键C-H1…Os1的原子轨道上也发生同样的现象。此外,Hs2,Hs3和O1轨道重叠存在不同的能量范围。这随后表明,当P-MAPTAC吸附在高岭石(001)表面上时,Hsn和 O1形成氢键,且键的轨道位置不同。
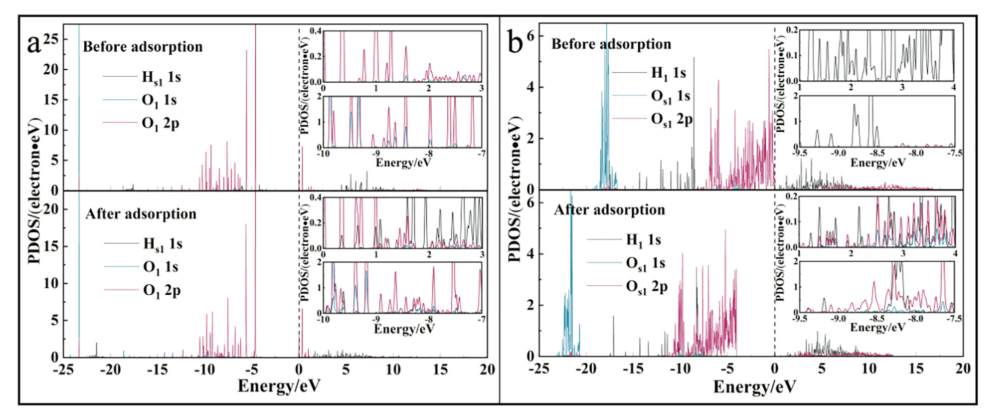
图7. P-DMC /高岭石(001)表面的键合原子的PDOS。费米能级用黑色虚线表示。
a:Hs1和O1,b:H1和Os1,拖影值为0.01。
解析:
在P-DMC /高岭石(001)表面的吸附系统中(图7),注意到Hs1和O1轨道重叠的能级主要位于-10 eV至-7 eV和0 eV至3 eV。从-9.5 eV到-7.5 eV和1 eV到4 eV的能量可以看出,Hs1轨道对Os1轨道起作用。此外,O1轨道负向移动时的能级不同于Os1,这证明了氢键(O-Hs1…O1)和氢键(C-H1…Os1)的键合性质之间存在差异。 PDOS用于比较高岭石表面上结构单元的氢键,以便对键合机理有更多的了解。
【总结】
利用DFT计算研究了三种CPAM结构单元在高岭石表面的吸附构型和机理。 CPAM结构单元的位置比高岭石表面的吸附位置对相互作用的影响更大。当吸附在高岭石(001)表面上时,P-DMDAAC的最佳吸附姿态为垂直姿态,而PMAPTAC和P-DMC为水平姿态。 P-DMDAAC呈垂直姿势,而P-MAPTAC和P-DMC呈倾斜姿势在高岭石(001(-))表面上形成最稳定的吸附构型。高岭石表面上结构单元的吸附能力顺序为P-MAPTAC> P-DMDAAC> P-DMC,而静电吸引力主导了整个吸附过程。在这项研究中,氢键O-Hsn…Om比C-HmOsn强。高岭石(001)表面的P-DMDAAC的结合本质是两个氢键(C-H1…Os1和C-H4…Os2),P-MAPTAC的氢键是O-Hsn…O1(n = 2和3)和C-H1…Os1和P-DMC具有两个氢键(C-H1…Os1和O-Hs1…O1)。当结构单元吸附在高岭石(001(-))表面上时,氢键吸附可能不稳定。氢键的性质导致原子轨道共振。即使氢键共享一个氧原子,轨道重叠的能量范围仍然不同。本研究为CPAM和高岭石的吸附机理提供了重要的见识,可为目标聚合单体的选择提供指导。 P-MAPTAC成为CPAM合成的重要选择。这项研究促进了理论和实验分析的结合,以解释使用PAM去除废水中的高悬浮固体。
全文链接:https://wvpn.ustc.edu.cn/https/77726476706e69737468656265737421e7e056d234336155700b8ca891472636a6d29e640e/science/article/pii/S0169131720302842
来源:
声明:仅代表作者个人观点,作者水平有限,如有不科学指出,欢迎指正


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









