






Introduction
Cancer is a global public health problem and the second most important cause of death in America (1). The global cancer burden is estimated every year by the American Cancer Society. According to the latest data report, the numbers of breast cancer (BC) cases and deaths estimated to occur in 2019 were 271,270 and 42,260, respectively (2). The high incidence and mortality of female BC remain a global health challenge, and the global burden is still increasing in several countries (3–5). Moreover, improvement of the overall clinical outcome of patients is crucial (6). Therefore, there is an urgent need to develop effective prognostic models for predicting the overall survival (OS) in patients with BC and for guiding clinical practice.
Metabolic reprogramming is a key hallmark of cancer (7, 8). Sufficient energy and metabolic intermediates for biosynthesis are the foundation of tumor cell initiation, proliferation and metastasis (9). Thus, many types of cancer are characterized by enhanced level of glycolysis and suppressed mitochondrial metabolism (7, 8, 10). Glycolysis might promote cancer cell survival by providing ATP and lactic acid (the main energy sources in cancer cells) (11). It has been reported that increased levels of glycolysis promoted the proliferation, invasion, and migration of certain cancer cells through activation of different signaling pathways and also enhanced drug resistance (10, 12–14). Therefore, tumor aerobic glycolysis has possible implications for prognosis judgment and cancer treatment (15, 16). Several studies have proven that the activity of cancer cells was significantly inhibited after glycolysis levels were decreased (17, 18). Studies have also examined the role of glycolysis in prediction of patient survival. For example, higher TCF7L2 expression predicted worse prognosis in pancreatic cancer (19). Four glycolysis-related genes (GRGs) (AGRN, AKR1A1, DDIT4, and HMMR) were identified as closely related to the clinical outcome in patients with lung adenocarcinoma (20). The glycolytic gene expression signatures based on nine (CLDN9, B4GALT1, GMPPB, B4GALT4, AK4, CHST6, PC, GPC1, and SRD5A3) and 10 biomarkers (HK2, HK3, LDHA, PKM2, GAPDH, ENO1, LDHB, PKLR, ALDOB, and GALM) predicted poor prognosis in patients with endometrial cancer (21) and glioblastoma patients (22), respectively. Although previous studies have investigated the role of GRGs and glycolysis in the development of BC (23–25), comprehensive investigations in this field are still needed.
This study aimed to evaluate the GRG expression in BC based on TCGA data and to study the association between GRG expression and BC survival. To this end, we primarily selected genes by conducting gene set enrichment analysis (GSEA). Many studies have focused on differentially expressed genes in tissues for the identification of biomarkers. However, some genes with important biological functions or connections among gene regulatory networks, gene functions, and characteristics are not differentially expressed and are often easily ignored. Since GSEA can scientifically screen genes based on the overall expression levels and data trends, it does not require significant differences in gene thresholds. This improves the statistical analysis of gene expression and biological significance (26).
Finally, an 11-GRG risk signature effectively predicting patient prognosis was constructed in our study. Furthermore, our gene-based model, as an independent prediction factor, could identify that patients with a high risk score had poorer prognoses than those in the low-risk score. Additionally, the prognosis performance of the risk model was significantly better than that of other clinical characteristics. In addition, it showed better performance in both training and testing datasets for predicting the clinical outcome in BC patients.
Materials and Methods
Data Collection
In total, 1,632 patients with BC were selected from five cohorts. The Cancer Genome Atlas (TCGA) cohorts included 1,057 records of patients with BC, whose expression profiles and clinical data were downloaded from the TCGA data portal (https://portal.gdc.cancer.gov/). The combined International Cancer Genomics Consortium (ICGC) cohort formed by the merger of Breast Cancer-FR and Breast Cancer-KR cohorts included 149 BC patients and the clinical information and expression profiles were obtained from ICGC database (http://dcc.icgc.org). Three Gene Expression Omnibus (GEO) cohorts were GSE42568, GSE7390, and GSE58812 datasets, which expression matrixes were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE58812 and GSE42568 expression profile was based on the GPL570 platform and respectively contained 107 and 121 BC samples (27, 28). The GSE7390 expression profile was based on the GPL96 platform, which cohort included 198 BC samples (29). The patients from TCGA were defined as a training cohort, while the four datasets from the ICGC and GEO were used for external validation. In addition, we also extracted detailed clinical information of TCGA cohort as shown in Table 1: age, pathological stage, estrogen receptor (ER; positive or negative) status, progesterone receptor (PR; positive or negative) status, human epidermal growth factor 2 (HER2; positive or negative) status, adjuvant chemotherapies, and T/N/M stage. GRG sets were searched from the Molecular Signatures Database (MSigDB) (30).
TABLE 1
Table 1 Clinic pathological characteristics of extracted patients with breast cancer.
Gene Set Enrichment Analysis
We used the GSEA (http://www.broadinstitute.org/gsea/index.jsp) to determine if the identified GRG sets had significant differences between the BC tissues and matched adjacent normal tissue (26). We use normalized P values of <0.05 to define statistical significance. The genes of the GRG sets which produced significant P value were collected for subsequent analysis.
Construction and Evaluation of the 11-GRG Prediction Model
We normalized each gene from among the expression profiles using log2 transformation (20, 31, 32). We sequentially conducted univariate Cox, the least absolute shrinkage and selection operator (LASSO) regression using the R package “glmnet” (33, 34), and multivariate Cox regression analyses to identify the GRGs associated with BC prognosis and to construct a GRG-based prediction model (34–37). The risk score was calculated using the following formula: Risk score = Σi=1ncoef∗id (38). We performed the Kaplan–Meier survival analysis to assess the difference in survival between high and low risk score groups by using “survival” R package (39, 40). The time-dependent receiver operating characteristic (ROC) curve was used to assess the performance of the gene risk model and compare the prediction efficiency with clinical features using the “survivalROC” R package (39). We applied Cox regression analyses to assess the independent prognostic values of the signature and other clinical characteristics. To estimate the likelihood of survival, a nomogram was constructed based on the risk score and clinical features by using the R package of “rms” (41), which were analyzed using multivariate Cox regression analysis. And the prognostic ability of the nomogram was weighed by C-index, ROC, and calibration plots (41).
Statistical Analysis
Differences among variables (risk score, age, tumor stage, T/N/M pathological stage, and ER, PR, and HER2 status) were tested using t-tests, non-parametric tests, or chi-square tests. We identified the alterations in selected genes from the cBioPortal website (http://www.cbioportal.org/). All statistical analyses were performed using R software (version 3.6.2) and R packages including “survivalROC”, “survival”, “glmnet” and “rms” (33, 34, 39, 40). P <0.05 was considered statistically different. All the scripts were uploaded at Github website (https://github.com/bioinformatics0/Glycolysis-BC).
Results
Initial Screening of Genes Using Gene Set Enrichment Analysis
We obtained a dataset containing clinical information on 1,057 BC patients and 112 normal controls and data on the expression levels of 24,991 mRNAs from TCGA. Five glycolysis-related MSigDB version 6.2 gene sets were downloaded, and a total of 443 genes were obtained. We used the above data and GSEA to verify which gene sets had significant differences between the BC tissues and matched adjacent normal tissues. The results demonstrated four significantly enriched gene sets, with normalized P values <0.05, from the following pathways: BIOCARTA_GLYCOLYSIS_PATHWAY, GO_GLYCOLYTIC_PROCESS, HALLMARK_GLYCOLYSIS, and REACTOME_ GLYCOLYSIS (Table 2, Figure 1). The corresponding 381 genes from these four gene sets were selected for subsequent analysis.
TABLE 2

Table 2 Gene sets enriched in breast cancer.
FIGURE 1

Figure 1 Enrichment plots of five gene sets which had significant difference between normal tissues and BC tissues by performing GSEA.
Construction and Evaluation of the Glycolysis-Related Risk Signature
We conducted univariate Cox regression analysis to analyze 381 genes after GSEA. Finally, a total of 11 genes (PGK1, SDC1, NUP43, NT5E, IL13RA1, GCLC, CACNA1H, P4HA1, TSTA3, MXI1, and STC1) were significantly correlated with OS (adjusted P < 0.05) after the filtration using LASSO and multivariable Cox regression analyses (Figure 2). A gene-based prognostic model was established to evaluate the survival risk for each patient as follows: Risk score = 0.00710 × expression of PGK1 + 0.00187 × expression of SDC1 + 0.05107 × expression of NUP43 + 0.05599 × expression of NT5E + 0.00587 × expression of IL13RA1 + 0.05692 × expression of GCLC + 0.01385 × expression of CACNA1H + (-0.00535) × expression of P4HA1 + 0.011698 × expression of TSTA3 + 0.026129 × expression of MXI1 + 0.00305 × expression of STC1. We then analyzed the mutational status of these 11 selected genes in TCGA BC samples in the cBioPortal database. Figure S1A shows the alterations in 11 genes. We also performed differential analysis of the expression of 11 genes in adjacent normal and BC tissues. Eleven genes were all significantly upregulated in tumor tissues (P < 0.05, Figure S1B).
FIGURE 2
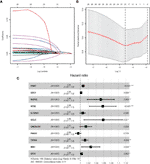
Figure 2 GRGs selection using the LASSO model and multivariable Cox model. (A) Ten-fold cross-validation for the coefficients of 326 GRGs in the LASSO model. (B) X-tile analysis of the 21 selected GRGs. (C) Forest plot illustrating the multivariable Cox model results of each gene in 11-GRG risk signature.
We calculated each patient’s risk scores in the training set based on the 11-gene signature. Patients with a high-risk score had a higher mortality rate than those with a low-risk score (P < 0.0001, log-rank test) (Figure 3A). The area under the curve (AUC) values for 1-, 3-, and 5-year OS, were 0.719, 0.762, and 0.742, respectively (Figure 3B). Figures 3C, D show the risk scores rank distribution and survival status in BC patients in the training set. The expression patterns of 11 GRGs in high/low risk groups are shown in the heatmap (Figure 3E). To assess the robustness of the 11-GRG signature, we assessed its performance using validation cohorts from the ICGC and GEO databases. Similar to that in the previous analysis, the patients in the high-risk subgroup had poorer survival than those in the low-risk group (P < 0.05; Figures 4Aa–Da). The 1-, 3-, and 5-year AUC values were 0.782, 0.79, and 0.675 in the ICGC cohort (Figure 4Ab), and 0.683, 0.723, and 0.752 in the GSE42568 cohort, respectively (Figure 4Bb). The AUC for OS was 0.715 at 1 year, 0.701 at 3 year and 0.76 at 5 year in the GSE7390 cohort (Figure 4Cb), and 0.711 at 1 year, 0.822 at 3 year, and 0.795 at 5 year in the GSE58812 cohort (Figure 4Db).
FIGURE 3

Figure 3 KM survival analysis, risk score assessment by the GRG‐related gene signature and time-dependent ROC curves in the TCGA cohort. (A) KM survival analysis of high‐ and low‐risk samples. (B) ROC curve for overall survival of the training set. The AUC was assessed at 1, 3, and 5 years. (C) Risk score distribution, (D), survival status, and (E). Eleven GRGs expression patterns for patients in high- and low-risk groups by the 11-GRG signature.
FIGURE 4
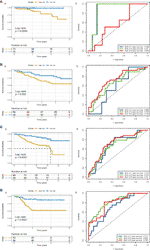
Figure 4 KM survival analysis and time-dependent ROC curves in the ICGC and GEO cohorts. (A) (ICGC), (B) a (GSE42568), (C) a (GSE7390), (D) a (GSE58812), Kaplan–Meier analysis with two-sided log-rank test was performed to estimate the differences in OS between the low-risk and high-risk group patients. (A) b (ICGC), (B) b (GSE42568), (C) b (GSE7390), (D) b (GSE58812), 1-, 3- and 5-year ROC curves of the 8-GRG signature were used to demonstrate the sensitivity and specificity in predicting the OS of BC patients.
Establishment and Assessment of a Nomogram
Univariate analyses were performed to examine the prognostic values of several clinicopathological features (age, pathological stage, ER, PR, and HER2). Consequently, the 11-GRG risk signature correlated with OS (hazard ration [HR] = 1.178; 95% confidence interval [CI], 1.128−1.231, P < 0.001) (Table 3). And age >60, (HR = 1.047; 95% CI, 1.030–1.064, P < 0.001), high pathological stage (III/IV) (HR = 2.022; 95% CI, 1.541−2.654, P < 0.001) were also risk factors for BC. Furthermore, after the multivariate analyses, the results showed that risk score (HR = 1.136; 95% CI, 1.083−1.191), age (HR = 1.047; 95% CI, 1.030–1.065), and stage (HR = 1.986; 95% CI, 1.522−2.591) remained independent prognostic factors with an adjusted P value <0.001. In addition, the ROC analysis revealed that the sensitivity and specificity of the 11-gene signature were greater than those of the other clinicopathological features (Figure 5A). Additionally, the gene risk model was proven to be a competitive prognostic factor for BC survival prediction. These results suggested that the signature can be a promising prognostic indicator for predicting OS in patients with BC. To develop a quantitative method that can predict the OS of patients with BC, a nomogram was constructed. The predictors included risk score, age, and tumor stage which produced significant P value in multivariate Cox analysis (Figure 5B). The result of C-index (0.812), AUC (1-year, 0.836; 3-year, 0.767 and 5-year, 0.792) and calibration plot showed the nomogram predicts with high accuracy (Figures 5C, D).
TABLE 3

Table 3 The risk score generated from the 11-GRG signature as an independent indicator according to Cox proportional hazards regression model.
FIGURE 5
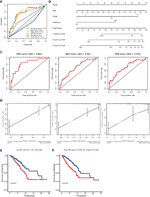
Figure 5 ROC curve with respect to clinical features and risk model, nomogram and Kaplan–Meier survival analysis for BC patients with clinical features: (A) Time-dependent ROC curve with respect to single clinical features and risk model. (B) The nomogram for predicting probabilities of BC patients overall survival. (C) 1-, 3- and 5-year ROC curves for the nomogram. (D) The 1-, 3- and 5-year nomogram calibration curves, respectively. Kaplan–Meier survival analysis for BC patients with different clinical features that can predict patient survival (E, Age, F, Stage).
Data Stratification Analyses
The results of the univariate Cox regression analysis of OS showed that age and stage could effectively predict survival in BC patients. The Kaplan–Meier curves revealed that the clinical features and results were consistent. BC patients who were older than 60 years and had stages III–IV disease were associated with poor prognosis (Figures 5E, F). In the TCGA cohort, subgroup analyses were conducted based on the clinicopathological variables (age, tumor stage, T/M/N stage, ER status, PR status, HER2 status and adjuvant chemotherapies). According to the Kaplan–Meier curves, in patients with BC who were stratified by age, tumor stage, T/N stage, ER status, PR status, HER2 status and adjuvant chemotherapies (No/Yes), the risk score remained a stable prognostic factor (Figures 6A–D, F–I). Nevertheless, the risk score played different roles in the subgroups or in patients stratified by metastasis stage. Patients in the high-risk group had a significantly shorter OS than those in the low-risk group in the subgroup of patients without distal metastasis (P < 0.001), while no significant difference was observed between the two groups with distal metastasis (P = 0.324) (Figure 6E). This result indicated that the risk model had better predictive value for clinical outcomes in BC patients without metastasis than in those with distal metastasis and more evidence and larger cohorts are necessary for further validation.
FIGURE 6
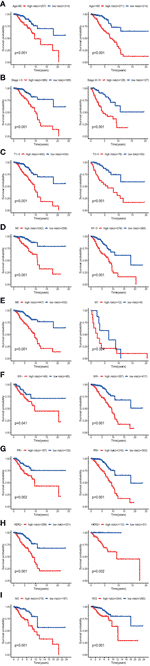
Figure 6 KM survival subgroup analysis of all patients with BC according to the GRG‐related gene signature stratified by clinical characteristics. (A) Age <60 y, Age >=60 y. (B) Early stage (stages I–II), Late stage (stages III–IV). C, T1-2, T3–4. (D) Lymph node-negative patients, Lymph node-positive patients. (E) Patients without distal metastasis, patients with distal metastasis. (F) ER-negative patients, ER-positive patients. (G) PR-negative patients, PR-positive patients. (H) HER2-negative patients, HER2-positive patients. (I) No adjuvant therapy, adjuvant therapy. GRGs, glycolysis-related genes; BC, breast cancer; ER, estrogen receptor; PR, progesterone receptor; HER2, human epidermal growth factor 2.
Comparison With Other Prognostic Signatures
A comparison of our nomogram and signature with other known prognostic hallmarks was performed. In order to exclude the impact of heterogeneity, all of these hallmarks that were developed based on TCGA database were included. Considering that our research is based on all types of BC and total TCGA BC cohort was used as the training set, so we further excluded the studies with the model construction for specific BC subtype (42–44) and studies which TCGA cohort was randomly divided into training and testing sets (45, 46). Finally, 15 related prognostic signatures were included to compare with our gene signature and nomogram (Table 4). The AUCs of the signature and the nomogram in our study at 1-, 3-, and 5-years were 0.719, 0.762, 0.742 and 0.836, 0.767, 0.792 respectively. Table 4 showed that the AUCs of four prognostic signature including 12 stemness-related lncRNA signature (0.813 at 5 years) (47), 11 immune-related lncRNA signature (0.836 at 5 years) (52), 27 immune-related gene signature (0.844 at 5 years) (54) and four methylated gene signature (0.791 at 5 years) (61) were distinctly higher than that of other biomarkers. Moreover, our signature also performed better in the prediction of BC patients’ OS than the signature based on the hallmarks related to autophagy (48), tumor microenvironment (immune, stromal, and proliferation) (49), tumor mutation burden (50), hypoxia (51), DNA repair (55), lncRNA (56) and miRNA (57, 58). The larger the AUC value of the biomarkers, the better the predictive ability of the hallmarks. This clearly shows that our nomogram and GRG signature are superior to other models after the four high-quality models in predicting the OS of BC patients.
TABLE 4

Table 4 The area under the ROC curve (AUC) show the sensitivity and specificity of the known signatures in predicting the prognosis of BC patients.
Discussion
BC is the most common cause of cancer-related mortality among malignancies and women worldwide (6, 62). It is difficult to predict prognosis in BC due to its phenotypic and molecular diversity. The application of prognostic models is useful for guiding clinical decisions and is essential for precision medicine. Subtype identification, risk stratification, and characterization of the underlying mechanisms are critical for the improvement of the existing treatment methods, development of more precise and personalized therapies, and prolongation of survival time. Glycolysis is a multi-step enzymatic reaction and is considered to be the root of the development and progression of cancer (63). Since an increasing number of studies have identified prognostic markers of GRGs, a GRG-based risk signature for predicting the survival in BC patients must be established to improve the accuracy in prognosis.
GSEA is a method for evaluating whole-genome expression profile chip data, which can integrate data from different levels and sources. In the present study, GSEA was conducted using the data on mRNA expression profiles in the 1,096 BC patients. Four gene sets with P values <0.05 exhibited significant differences and were chosen for subsequent analyses. Univariate, multivariate Cox, and LASSO regression analyses were performed to identify 11 prognostic genes for BC patients. Based on the 11 most valuable biomarkers, we developed and verified an effective model to predict clinical outcomes in BC patients. Survival analysis showed distinctly different prognoses between high- and low-risk BC patients. The model was also verified in the GEO and ICGC datasets, demonstrating favorable clinical predictive ability. In addition, the prediction model for BC patients could act as an independent prognostic tool through multivariate Cox analyses. We also found that patients with higher risk scores in our prediction model tended to be older, have advanced stage disease, and a poorer prognosis. The prediction model in our study had similar or better clinical application potential compared to traditional clinical factors. Moreover, we integrated the prediction model and clinical characteristics to establish a novel nomogram. The nomogram took advantage of the complementary values of clinical characteristics and the prediction model and provided superior estimation of OS. The result showed that C-index, ROC and calibration plot performed well in our study. Additionally, the gene signature could further stratify clinically defined groups of patients (e.g., groups stratified according to age, stage, T/N/M stage, ER status, PR status, HER2 status and adjuvant chemotherapies) into subgroups with different survival outcomes. The risk model could effectively predict the prognosis of patients with BC in all subgroups, but it could not be applied to the subgroup of BC patients with distal metastasis. The underlying mechanisms of this result should be explored in depth in the future. The results showed that the calculation of risk scores has great prognostic significance for BC patients. This not only increases the means of predicting the prognosis but can also help clinicians to choose more suitable treatment options for patients.
Chemotherapy is still an important way of cancer treatment. Chemotherapy drugs have an oxygen-dependent effect on the killing of tumor cells, most of which kill cells by oxidizing free radicals and reactive oxygen species in cells. Hypoxia can significantly reduce the efficiency of chemotherapy (64). The Warburg effect is aerobic glycolysis in cancer cells, which has been found to be involved in chemotherapy resistance in various types of human cancers (65, 66). The Warburg effect promotes epigenetic and genetic changes leading to the occurrence of multiple new cell phenotypes including the existence of drug resistance cells (67). To confirm whether our signature can provide an effective prediction method for the prognosis of patients receiving adjuvant chemotherapy, we conducted a subgroup analysis and the results showed the risk model could effectively predict the prognosis of patients with BC in both receiving and not receiving adjuvant chemotherapy groups. This also shows the extensive clinical application of our model.
To further explore the predictive ability of our nomogram, a comparison was performed among several significant molecular signatures which were employed for predicting OS of BC patients. The studies (47, 48, 52, 54, 61) we included were that the model was built based on the entire TCGA cohort and involved all types of breast cancer, not a certain subtype. The final results showed that our signature and another four prognostic signature including 12 stemness-related lncRNA signature (47), 11 immune-related lncRNA signature (52), 27 immune-related gene signature (54) and four methylated gene signature (61) performed better in the prediction of BC patients’ OS than the signature based on the hallmarks related to autophagy (48), tumor microenvironment (immune, stromal, and proliferation) (49), tumor mutation burden (50), hypoxia (51), DNA repair (55), lncRNA (56) and miRNA (57, 58). Considering that the clinical application cost of our model may be lower than that of the two gene models [12 stemness-related lncRNA signature (47) and 27 immune-related gene signature (54)] and glycolysis is closely related to the prognosis of BC, our signature may be necessary to enrich the clinical prediction methods. What’s more, the AUC of the nomogram is greater than that of the signature in our study, suggesting that the combination of the risk score with clinical factors is more promising than the methylation signature alone in predicting the OS of BC patients.
The 11 GRGs identified in this study included PGK1, SDC1, NUP43, NT5E, IL13RA1, GCLC, CACNA1H, P4HA1, TSTA3, MXI1, and STC1. Of these genes, PGK1 (phosphoglycerate kinase 1) has been identified to promote BC progression and metastases via forming a positive feed-forward loop with HIF-1α. High PGK1 expression predicted poor prognosis in BC (63). SDC1 (syndecan-1), a heparin cell surface proteoglycan, can act as a co-receptor for growth factors and chemokines (68). High expression of SDC1 has been identified in BC tissues as associated with an aggressive phenotype and poor clinical behavior (69). Nup43 (nucleoporin 43 kDa) is a stable component of the Nup107 160 complex, which is localized at kinetochores in mitosis and regulates mitotic progression and chromosome segregation (70). Higher expression of NUP43 is often accompanied by DNA amplification and is related to poor OS in luminal A and HER2+ BC (71). NT5E (ecto-5-nucleotidase), also designated CD73, is a promising prognostic factor, and its high expression was significantly related to lymph node metastases in BC patients (72). A study reported that the interactions between interleukin-13 and interleukin-13 receptor alpha 1 (IL13RA1) contributed to cancer cell growth and metastasis, and IL13RA1 expression was associated with poor prognosis in invasive BC patients (73). Collagen prolyl 4-hydroxylase alpha 1 (P4HA1) is the major isoform in most cell types and tissues, and it can also enhance the activity of most prolyl 4-hydroxylases (74). During the development of BC, P4HA1 expression is induced (75). When the P4HA/HIF-1 axis is activated, the cancer cell stemness is enhanced, while the levels of oxidative phosphorylation and reactive oxygen species are reduced (76). The malignant transformation of cells and tumor development were promoted by abnormal glycosylation, which depends on TSTA3 gene function (tissue-specific transplantation antigen P35B) (77). The survival rates in BC patients with a higher expression of TSTA3 were lower (78). MYC-associated protein X interactor-1 (MXI1) is an antagonist of the oncogenic MYC protein, and the deletion of the MXI1 gene causes many kinds of human cancers (79). The low expression of MXI1 was related to poor prognosis in BC patients (80). Stanniocalcin-1 (STC1) is a secreted glycoprotein, and its high expression levels were associated with tumor growth and metastasis in BC (81). However, other genes (GCLC and CACNA1H) were identified for the first time to have prognostic value in BC patients. Deeper investigations of the biological functions of these genes in BC are warranted.
To our knowledge, our study is the first one to identify and comprehensively analyze prognostic GRGs for the prediction of survival in BC patients by evaluating the data from the public TCGA database. Moreover, a novel risk signature based on 11 GRGs was identified and verified. This signature can be used as a screening tool for patients at high risk of developing BC and to stratify patients to increase the effectiveness of targeted therapy. Additionally, we successfully established a GRG-related nomogram combining clinical factors and molecular markers to predict the OS of BC patients with in an effective quantitative approach. We also analyzed the mutational status of the nine selected genes in the cBioPortal database. Our research not only allowed to better understand the genetics of BC, but also had significance for guiding future research.
There are some limitations in our study. First, it was a retrospective study, and all BC patients were identified from public databases. Second, large-scale multicenter cohorts are necessary to validate the predictive performance of our model and to evaluate its clinical applicability for better management of BC. Furthermore, future basic experiments in our hospital will be required to conduct to verify our findings and elucidate the functional roles of GRGs involved in the initiation and development of BC. Moreover, the gene signature may be more effective to predict survival in BC patients without distal metastasis, and its prognostic role warrants further evaluation.
Conclusion
We constructed a valid, innovative, and reliable 11-GRGs prognostic model (PGK1, SDC1, NUP43, NT5E, IL13RA1, GCLC, CACNA1H, P4HA1, TSTA3, MXI1, and STC1) to predict BC patient outcomes. Our signature was an independent and important risk factor for BC. Furthermore, a nomogram combining the prediction model and clinical factors was constructed, which could be a useful tool to predict prognosis and guide clinical practice.
Data Availability Statement
The datasets generated and analyzed during the current study are available in the TCGA (http://cancergenome.nih.gov/abouttcga), GEO (https://www.ncbi.nlm.nih.gov/geo/) ICGC (http://dcc.icgc.org) and cBioPortal (http://www.cbioportal.org) databases. Accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the First Affiliated Hospital of Zhejiang University in Zhejiang Province (Hangzhou, China). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
DZ and YiZ collected and analyzed the data, and wrote the manuscript. SY, YL, MW, and JY analyzed the data and reviewed the manuscript. YD, NL, BW, YuZ, and YW participated in analyzing the data. DZ, YiZ, SY, and YL participated in the preparation of the figures and tables and interpretation of data for the work. ZD and HL designed the work and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank The Cancer Genome Atlas (TCGA) project, NCBI GEO, and cBioPortal database and their contributors for these valuable public data sets.
Supplementary Material
Supplementary Figure 1 | Identification of GRGs related to patients’ survival. (A) Selected genes’ alteration in TCGA samples. (B) Different expression of eleven selected genes (*P < 0.05, **P < 0.01 and ***P < 0.001).
Abbreviations
BC, Breast cancer; ER, Estrogen receptor; GEO, Gene Expression Omnibus; GRG, Glycolysis-related gene; GSEA, Gene set enrichment analysis; ICGC, International Cancer Genomics Consortium; LASSO, Least absolute shrinkage and selection operator; OS: Overall survival; PR, Progesterone receptor; ROC, Receiver operating characteristic; TCGA, The Cancer Genome Atlas.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
2. Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C, et al. European cancer mortality predictions for the year 2019 with focus on breast cancer. Ann Oncol (2019) 30(5):781–7. doi: 10.1093/annonc/mdz051
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
4. Coughlin SS, Ekwueme DU. Breast cancer as a global health concern. Cancer Epidemiol (2009) 33(5):315–8. doi: 10.1016/j.canep.2009.10.003
5. Biglia MDA N, Sgro LG, Tomasi Cont N, Bounous V, Robba E. BREAST CANCER TREATMENT IN MUTATION CARRIERS. Minerva Ginecol (2016) 68(5):548–56.
6. Irun R, Cohen AL, Lambris JD, Paoletti R, Rezaei N. Breast Cancer Metastasis and Drug Resistance Challenges and Progress. Adv Exp Med Biol (2019).
7. Cassim S, Vučetić M, Ždralević M, Pouyssegur J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers (Base) (2020) 12(5):1119. doi: 10.3390/cancers12051119
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
9. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7(1):11–20. doi: 10.1016/j.cmet.2007.10.002
10. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer (2013) 12:152. doi: 10.1186/1476-4598-12-152
11. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi: 10.1038/nrc3038
12. Bhattacharya D, Scimè A. Metabolic Regulation of Epithelial to Mesenchymal Transition: Implications for Endocrine Cancer. Front Endocrinol (2019) 10:773. doi: 10.3389/fendo.2019.00773
13. Li C, Zhang G, Zhao L, Ma Z, Chen H. Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J Surg Oncol (2016) 14(1):15. doi: 10.1186/s12957-016-0769-9
14. Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S, et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun (2017) 8:14634. doi: 10.1038/ncomms14634
15. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
16. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Sci (N Y NY) (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
17. Robinson GL, Dinsdale D, Macfarlane M, Cain K. Switching from aerobic glycolysis to oxidative phosphorylation modulates the sensitivity of mantle cell lymphoma cells to TRAIL. Oncogene (2012) 31(48):4996–5006. doi: 10.1038/onc.2012.13
18. Kim SM, Yun MR, Hong YK, Solca F, Kim J-H, Kim H-J, et al. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol Cancer Ther (2013) 12(10):2145–56. doi: 10.1158/1535-7163.mct-12-1188
19. Xiang J, Hu Q, Qin Y, Ji S, Xu W, Liu W, et al. TCF7L2 positively regulates aerobic glycolysis via the EGLN2/HIF-1α axis and indicates prognosis in pancreatic cancer. Cell Death Dis (2018) 9(3):321. doi: 10.1038/s41419-018-0367-6
20. Liu C, Li Y, Wei M, Zhao L, Yu Y, Li G. Identification of a novel glycolysis-related gene signature that can predict the survival of patients with lung adenocarcinoma. Cell Cycle (Georgetown Tex) (2019) 18(5):568–79. doi: 10.1080/15384101.2019.1578146
21. Wang ZH, Zhang YZ, Wang YS, Ma XX. Identification of novel cell glycolysis related gene signature predicting survival in patients with endometrial cancer. Cancer Cell Int (2019) 19:296. doi: 10.1186/s12935-019-1001-0
22. Chen C, Shi Y, Li Y, He ZC, Zhou K, Zhang XN, et al. A glycolysis-based ten-gene signature correlates with the clinical outcome, molecular subtype and IDH1 mutation in glioblastoma. J Genet Genomics = Yi Chuan xue bao (2017) 44(11):519–30. doi: 10.1016/j.jgg.2017.05.007
23. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene (2018) 37(8):1062–74. doi: 10.1038/onc.2017.368
24. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab (2018) 28(1):87–103. doi: 10.1016/j.cmet.2018.04.022
25. Xing Z, Zhang Y, Liang K, Yan L, Xiang Y, Li C, et al. Expression of Long Noncoding RNA YIYA Promotes Glycolysis in Breast Cancer. Cancer Res (2018) 78(16):4524–32. doi: 10.1158/0008-5472.can-17-0385
26. Thomas MA, Yang L, Carter BJ, Klaper RD. Gene set enrichment analysis of microarray data from Pimephales promelas (Rafinesque), a non-mammalian model organism. BMC Genomics (2011) 12:66. doi: 10.1186/1471-2164-12-66
27. Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, O’Driscoll L, et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis (2013) 34(10):2300–8. doi: 10.1093/carcin/bgt208
28. Jézéquel P, Loussouarn D, Guérin-Charbonnel C, Campion L, Vanier A, Gouraud W, et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response. Breast Cancer Res BCR (2015) 17:43. doi: 10.1186/s13058-015-0550-y
29. Patil P, Bachant-Winner PO, Haibe-Kains B, Leek JT. Test set bias affects reproducibility of gene signatures. Bioinf (Oxford Engl) (2015) 31(14):2318–23. doi: 10.1093/bioinformatics/btv157
30. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinf (Oxford Engl) (2011) 27(12):1739–40. doi: 10.1093/bioinformatics/btr260
31. Chen K, He Y, Liu Y, Yang X. Gene signature associated with neuro-endocrine activity predicting prognosis of pancreatic carcinoma. Mol Genet Genomic Med (2019) 7(7):e00729. doi: 10.1002/mgg3.729
32. Zhang L, Zhang Z, Yu Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J Trans Med (2019) 17(1):423. doi: 10.1186/s12967-019-02173-2
33. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw (2010) 33(1):1–22. doi: 10.18637/jss.v033.i01
34. Wang H, Lengerich BJ, Aragam B, Xing EP. Precision Lasso: accounting for correlations and linear dependencies in high-dimensional genomic data. Bioinf (Oxford Engl) (2019) 35(7):1181–7. doi: 10.1093/bioinformatics/bty750
35. Tibshirani R. THE LASSO METHOD FOR VARIABLE SELECTION IN THE COX MODEL. Stat Med (1997) 16(4):385–95. doi: 10.1002/(sici)1097-0258(19970228)16:4<385::aid-sim380>3.0.co;2-3
36. Cao R, Wu Q, Li Q, Yao M, Zhou H. A 3-mRNA-based prognostic signature of survival in oral squamous cell carcinoma. PeerJ (2019) 7:e7360. doi: 10.7717/peerj.7360
37. Yang S, Wu Y, Deng Y, Zhou L, Yang P, Zheng Y, et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology (2019) 8(12):e1659094. doi: 10.1080/2162402x.2019.1659094
38. Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med (2007) 356(1):11–20. doi: 10.1056/NEJMoa060096
39. Lorent M, Giral M, Foucher Y. Net time-dependent ROC curves: a solution for evaluating the accuracy of a marker to predict disease-related mortality. Stat Med (2014) 33(14):2379–89. doi: 10.1002/sim.6079
40. Harrell FE Jr., Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med (1996) 15(4):361–87. doi: 10.1002/(sici)1097-0258(19960229)15:4<361::aid-sim168>3.0.co;2-4
41. Ma X, Cheng J, Zhao P, Li L, Tao K, Chen H. DNA methylation profiling to predict recurrence risk in stage I lung adenocarcinoma: Development and validation of a nomogram to clinical management. J Cell Mol Med (2020) 24(13):7576–89. doi: 10.1111/jcmm.15393
42. Tian T, Gong Z, Wang M, Hao R, Lin S, Liu K, et al. Identification of long non-coding RNA signatures in triple-negative breast cancer. Cancer Cell Int (2018) 18:103. doi: 10.1186/s12935-018-0598-8
43. Xiao B, Chen L, Ke Y, Hang J, Cao L, Zhang R, et al. Identification of methylation sites and signature genes with prognostic value for luminal breast cancer. BMC Cancer (2018) 18(1):405. doi: 10.1186/s12885-018-4314-9
44. Li C, Li X, Li G, Sun L, Zhang W, Jiang J, et al. Identification of a prognosis−associated signature associated with energy metabolism in triple−negative breast cancer. Oncol Rep (2020) 44(3):819–37. doi: 10.3892/or.2020.7657
45. He Y, Li X, Meng Y, Fu S, Cui Y, Shi Y, et al. A prognostic 11 long noncoding RNA expression signature for breast invasive carcinoma. J Cell Biochem (2019) 120(10):16692–702. doi: 10.1002/jcb.28927
46. Wang H, Wu L, Wang H. Development and verification of a personalized immune prognostic feature in breast cancer. Exp Biol Med (Maywood NJ) (2020) 245(14):1242–53. doi: 10.1177/1535370220936964
47. Li X, Li Y, Yu X, Jin F. Identification and validation of stemness-related lncRNA prognostic signature for breast cancer. J Trans Med (2020) 18(1):331. doi: 10.1186/s12967-020-02497-4
48. Lin QG, Liu W, Mo YZ, Han J, Guo ZX, Zheng W, et al. Development of prognostic index based on autophagy-related genes analysis in breast cancer. Aging (Albany NY) (2020) 12(2):1366–76. doi: 10.18632/aging.102687
49. Wang J, Yang Z, Zhang C, Ouyang J, Zhang G. Wu C. A four-gene signature in the tumor microenvironment that significantly associates with the prognosis of patients with breast cancer. Gene (2020) 761:145049. doi: 10.1016/j.gene.2020.145049
50. Wang F, Tang C, Gao X, Xu J. Identification of a six-gene signature associated with tumor mutation burden for predicting prognosis in patients with invasive breast carcinoma. Ann Trans Med (2020) 8(7):453. doi: 10.21037/atm.2020.04.02
51. Wang J, Wang Y, Xing P, Liu Q, Zhang C, Sui Y, et al. Development and validation of a hypoxia-related prognostic signature for breast cancer. Oncol Lett (2020) 20(2):1906–14. doi: 10.3892/ol.2020.11733
52. Shen Y, Peng X, Shen C. Identification and validation of immune-related lncRNA prognostic signature for breast cancer. Genomics (2020) 112(3):2640–6. doi: 10.1016/j.ygeno.2020.02.015
53. Xu H, Wang G, Zhu L, Liu H, Li B. Eight immune-related genes predict survival outcomes and immune characteristics in breast cancer. Aging (Albany NY) (2020) 12(16):16491–513. doi: 10.18632/aging.103753
54. Zhao Y, Pu C, Liu Z. Exploration the Significance of a Novel Immune-Related Gene Signature in Prognosis and Immune Microenvironment of Breast Cancer. Front Oncol (2020) 10:1211. doi: 10.3389/fonc.2020.01211
55. Zhang D, Yang S, Li Y, Yao J, Ruan J, Zheng Y, et al. Prediction of Overall Survival Among Female Patients With Breast Cancer Using a Prognostic Signature Based on 8 DNA Repair-Related Genes. JAMA Netw Open (2020) 3(10):e2014622. doi: 10.1001/jamanetworkopen.2020.14622
56. Sun M, Wu D, Zhou K, Li H, Gong X, Wei Q, et al. An eight-lncRNA signature predicts survival of breast cancer patients: a comprehensive study based on weighted gene co-expression network analysis and competing endogenous RNA network. Breast Cancer Res Treat (2019) 175(1):59–75. doi: 10.1007/s10549-019-05147-6
57. Kawaguchi T, Yan L, Qi Q, Peng X, Edge SB, Young J, et al. Novel MicroRNA-Based Risk Score Identified by Integrated Analyses to Predict Metastasis and Poor Prognosis in Breast Cancer. Ann Surg Oncol (2018) 25(13):4037–46. doi: 10.1245/s10434-018-6859-x
58. Lai J, Wang H, Pan Z, Su F. A novel six-microRNA-based model to improve prognosis prediction of breast cancer. Aging (Albany NY) (2019) 11(2):649–62. doi: 10.18632/aging.101767
59. Liu L, Chen Z, Shi W, Liu H, Pang W. Breast cancer survival prediction using seven prognostic biomarker genes. Oncol Lett (2019) 18(3):2907–16. doi: 10.3892/ol.2019.10635
60. Tao C, Luo R, Song J, Zhang W, Ran LA seven-DNA methylation signature as a novel prognostic biomarker in breast cancer. J Cell Biochem (2020) 121(3):2385–93. doi: 10.1002/jcb.29461
61. Feng L, Jin F. Screening of differentially methylated genes in breast cancer and risk model construction based on TCGA database. Oncol Lett (2018) 16(5):6407–16. doi: 10.3892/ol.2018.9457
62. DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin (2019) 69(3):211–33. doi: 10.3322/caac.21555
63. Fu D, He C, Wei J, Zhang Z, Luo Y, Tan H, et al. PGK1 is a Potential Survival Biomarker and Invasion Promoter by Regulating the HIF-1α-Mediated Epithelial-Mesenchymal Transition Process in Breast Cancer. Cell Physiol Biochem (2018) 51(5):2434–44. doi: 10.1159/000495900
64. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18(1):157. doi: 10.1186/s12943-019-1089-9
65. He M, Jin Q, Chen C, Liu Y, Ye X, Jiang Y, et al. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene (2019) 38(28):5551–65. doi: 10.1038/s41388-019-0817-3
66. Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol (2016) 173(6):970–9. doi: 10.1111/bph.13422
67. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updates (2018) 38:1–11. doi: 10.1016/j.drup.2018.03.001
68. Shi S, Zhong D, Xiao Y, Wang B, Wang W, Zhang F, et al. Syndecan-1 knockdown inhibits glioma cell proliferation and invasion by deregulating a c-src/FAK-associated signaling pathway. Oncotarget (2017) 8(25):40922–34. doi: 10.18632/oncotarget.16733
69. Cui X, Jing X, Yi Q, Long C, Tian J, Zhu J. Clinicopathological and prognostic significance of SDC1 overexpression in breast cancer. Oncotarget (2017) 8(67):111444–55. doi: 10.18632/oncotarget.22820
70. Loïodice I, Alves A, Rabut G, Van Overbeek M, Ellenberg J, Sibarita JB, et al. The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell (2004) 15(7):3333–44. doi: 10.1091/mbc.e03-12-0878
71. Tian C, Zhou S, Yi C. High NUP43 expression might independently predict poor overall survival in luminal A and in HER2+ breast cancer. Future Oncol (Lond Engl) (2018) 14(15):1431–42. doi: 10.2217/fon-2017-0690
72. Jiang T, Xu X, Qiao M, Li X, Zhao C, Zhou F, et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer (2018) 18(1):267. doi: 10.1186/s12885-018-4073-7
73. Park MH, Kwon HJ, Kim JR, Lee B, Lee SJ, Bae YK. Elevated Interleukin-13 Receptor Alpha 1 Expression in Tumor Cells Is Associated with Poor Prognosis in Patients with Invasive Breast Cancer. Ann Surg Oncol (2017) 24(12):3780–7. doi: 10.1245/s10434-017-5907-2
74. Annunen P, Autio-Harmainen H, Kivirikko KI. The novel type II prolyl 4-hydroxylase is the main enzyme form in chondrocytes and capillary endothelial cells, whereas the type I enzyme predominates in most cells. J Biol Chem (1998) 273(11):5989–92. doi: 10.1074/jbc.273.11.5989
75. Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res (2013) 73(11):3285–96. doi: 10.1158/0008-5472.can-12-3963
76. Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun (2018) 9(1):4456. doi: 10.1038/s41467-018-06893-9
77. Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res (2008) 68(10):3803–9. doi: 10.1158/0008-5472.can-07-6389
78. Sun Y, Liu X, Zhang Q, Mao X, Feng L, Su P, et al. Oncogenic potential of TSTA3 in breast cancer and its regulation by the tumor suppressors miR-125a-5p and miR-125b. Tumour Biol (2016) 37(4):4963–72. doi: 10.1007/s13277-015-4178-4
79. Cascón A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res (2012) 72(13):3119–24. doi: 10.1158/0008-5472.can-11-3891
80. Xu LP, Sun Y, Li W, Mai L, Guo YJ, Fan QX. MYC and MXI1 protein expression: potential prognostic significance in women with breast cancer in China. Oncol Res Treat (2014) 37(3):118–23. doi: 10.1159/000360207
81. Chang AC, Doherty J, Huschtscha LI, Redvers R, Restall C, Reddel RR, et al. STC1 expression is associated with tumor growth and metastasis in breast cancer. Clin Exp Metastasis (2015) 32(1):15–27. doi: 10.1007/s10585-014-9687-9















 3万+
3万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









