






大家好!为大家分享本课题组近期发表在Nucleic Acids Research的文章,题目为 “Entropy subspace separation-based clustering for noise reduction (ENCORE) of scRNA-seq data”,文章提出了一种基于表达密度谱的特征选择方法,能够有效进行特征子空间分离完成特征信息和噪音识别,并结合一致性分群策略,设计了一种单细胞转录组分群新算法(ENCORE)。

【背景介绍】
单细胞转录组测序技术使得我们能够捕获单个细胞的转录组信息,为复杂生物问题的研究提供更精细的分析手段。为了更有效地利用单细胞转录组数据、提取细胞异质性信息,我们需要使用高精度和高分辨率的聚类相关算法,因此近年来研究者们已经开发了大量单细胞转录组分群算法。这些算法通常包括归一化、特征选择、降维、距离计算、聚类、分群标记基因识别等关键步骤,新的算法也常是针对以上步骤进行改进。然而相比于其他步骤,特征选择的改进发展较为缓慢。多数算法选择在表达上具有高表达、高变异性的特征(基因或转录本)进行下游分析,该类方法容易受到噪音的干扰而丢失有效特征,导致结果被高表达的特征主导。
【设计思路】为了解决目前特征选择所存在的问题,ENCORE提出了一种独特的子空间分离策略,用于降噪和特征选择,从而实现单细胞分群的优化。ENCORE的设计基于这样一个假设:在不考虑表达量的情况下,细胞间表达密度谱相似的特征可能携带相似的细胞异质性信息,因此将特征根据密度谱进行子空间分离后,细胞群在这些子空间中可能呈现更清晰的分布。具体过程如图1所示,主要包括子空间分离、子空间内分群、一致性分群三个步骤。首先对特征的表达密度谱进行分群,将对应特征分离到不同的子空间,这些子空间在各个维度上包含相当的异质性信息,因而更有利于提取异质性信息。随后ENCORE通过识别细胞簇来评估子空间的“熵”值,即子空间包含异质性信息的丰富程度。低熵子空间具有清晰的分群信息,细胞的分布规则;高熵子空间不具有清晰的分群信息,细胞的分布随机。接着ENCORE通过对低熵子空间的筛选实现更有效、准确的特征选择,该方法不仅能保留低表达的有效特征,而且对相似异质性特征的分群使得聚类信号更加集中。最后,ENCORE又利用新设计的一致性分群算法,实现了不同子空间分群信息的整合,增强了来自多个低熵子空间的共同信号并保留了各子空间的特有信号。在这种设计框架下,ENCORE可以在不同的单细胞转录组数据集上实现精确的细胞分群、二维可视化以及分群标记基因识别。
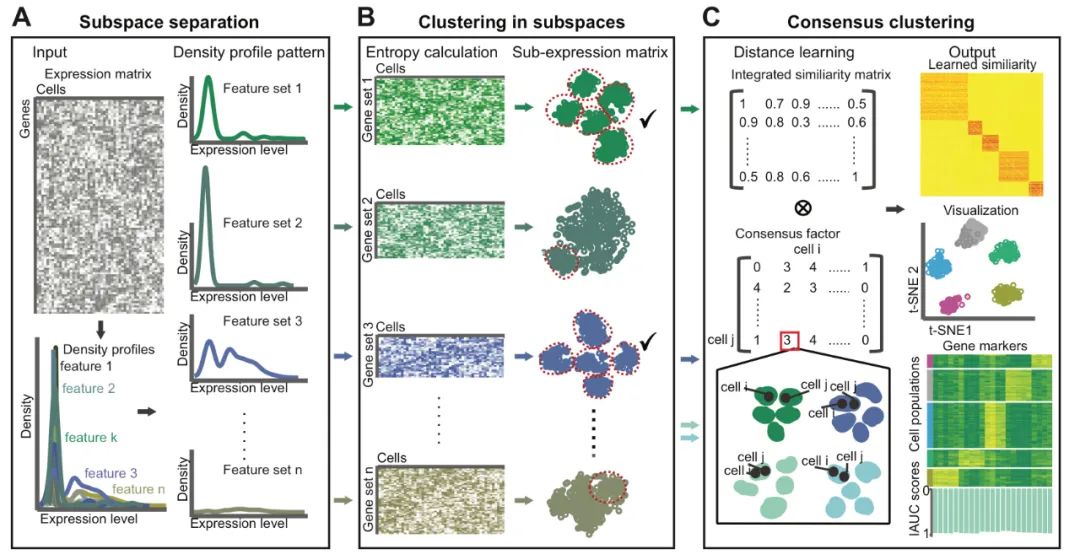
图1. ENCORE算法示意图
【数据介绍】首先我们将ENCORE应用于12个标准数据集和浙大小鼠细胞图谱的大数据集,以评估ENCORE的子空间分离性能以及特征选择、降噪能力。从图2可以看出,无论是小数据集(图2A)还是大数据集(图2B),ENCORE的子空间分离方法均能生成多个具有不同信息量的子空间。其中图1A的子空间2,3,4呈现较规则的细胞群分布,子空间1的细胞群分布则相对不规则;图1B的子空间34和43相较于其余子空间也具有较好的分群信息,说明ENCORE可以识别小数据集(图2A)或大数据集(图2B)的低熵子空间。图3展示了Seurat与ENCORE特征选择的结果比较,ENCORE选取的特征在变异系数和均值上具有更随机的分布,说明其与传统算法在特征选择上存在较大差异。

图2. ENCORE子空间分离效果
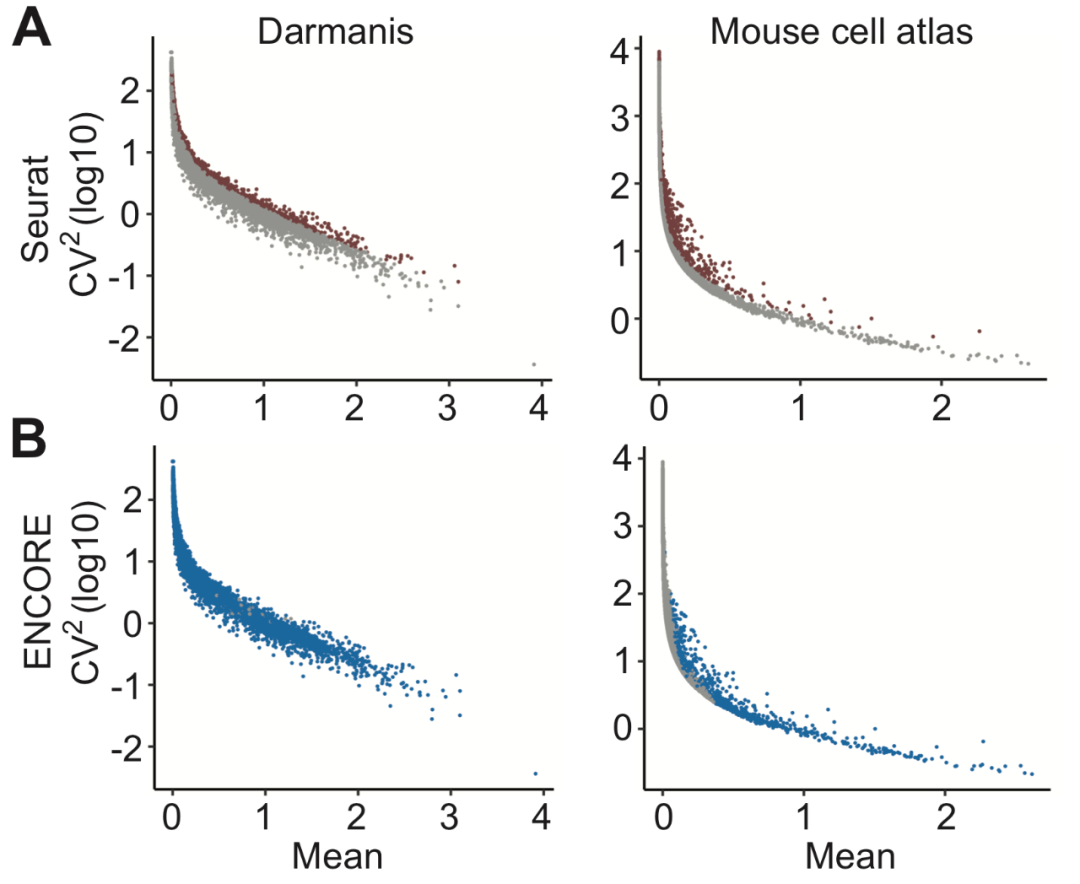
图3. ENCORE与Seurat特征选择结果比较
接着,通过与四种广泛应用且高集成性的单细胞分群算法(Seurat, SIMLR, pcaReduce and t-SNE + K-means)进行比较,ENCORE验证了其分群的准确性和普适性。以12个具有已知实验分群标签的标准数据集作为测试数据集,并使用ARI(Adjusted Rand Index)和NMI(Normalized Mutual Information)作为分群准确性指标,两个指标的值越高说明算法预测分群与已知分群标签具有越好的一致性,即说明算法预测分群的准确性越高。如图4A所示,相比于其他算法,在所有数据集上,ENCORE都表现出最高或相当的分群准确性,即使是细胞数较少的数据集,ENCORE仍具有最好的分群准确性。由于这12个数据集来自不同测序平台,具有不同的数据量大小和测序深度,并且使用了不同的归一化方法,而ENCORE都表现出较好的分群准确性,说明ENCORE的分群性能更为稳健。同时,相较于其他算法,ENCORE的可视化结果不仅与聚类结果具有高度的一致性,而且更为清晰、直观(图4B)。
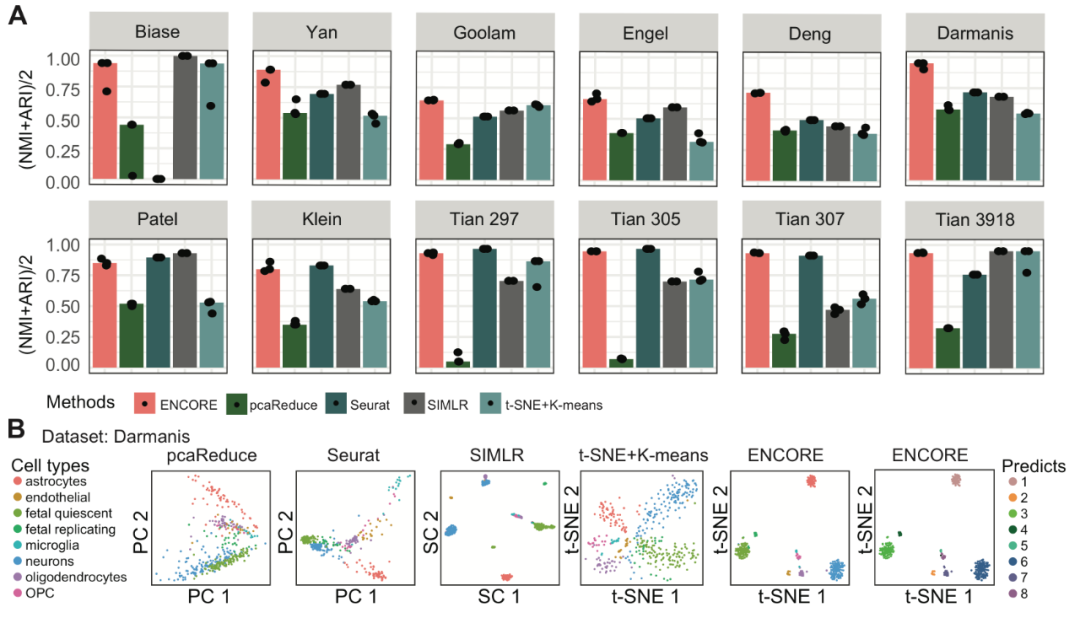
图4. ENCORE与其他算法的准确性及可视化结果比较
最后,为了评估了ENCORE对难分离数据集的信息提取能力,我们使用ENCORE对一个小鼠脂肪祖细胞数据集进行分析(图5)。尽管脂肪祖细胞间存在异质性,但是从单细胞转录组数据中识别清晰的分群和明显的标记基因还是困难的。从结果可以看出,ENCORE能够产生更清晰的细胞分型,并识别更具特异性的亚型标记基因,这些亚型标记基因的表达在热图上也能显示出清晰的模式(图5B)。接着,我们挑选了亚型标记基因Mgp(该基因为ENCORE推导的group 8的最优标志基因),对其功能展开初步探索。我们发现3T3-L1细胞中Mgp基因的过表达并不会影响成脂分化相关基因的表达,但是会显著提高Dio2基因的表达(图5D)。Dio2蛋白与甲状腺素(T4)向三碘甲状腺原氨酸(T3)转化相关。分析显示Mgp能够上调T3浓度,而又有报道显示T3能够刺激Mgp的表达上调,说明T3和Mgp之间存在潜在的正反馈回路(图5E)。这些结果表明,ENCORE能够从复杂的数据集中识别具有生物学意义的标记信息。
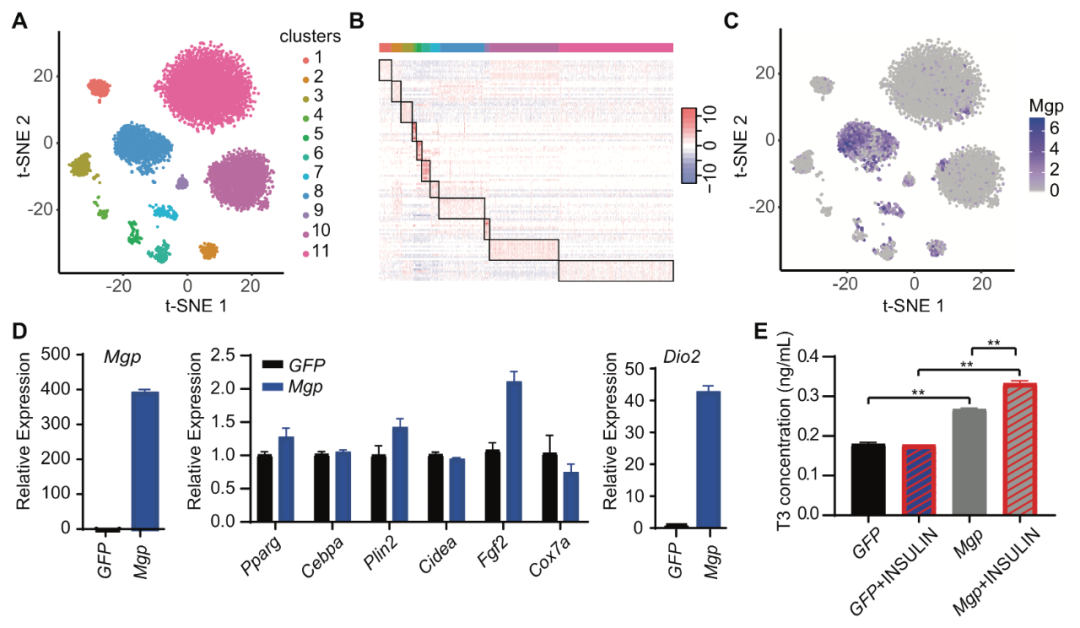
图5. ENCORE在小鼠脂肪祖细胞分群中的应用
【总结】在这篇文章中,本课题组和复旦大学李晋课题组开发了一种新型scRNA-seq分析方法ENCORE。基于相似表达密度谱的特征具有相似异质性信息的假设,ENCORE将特征分到不同的熵子空间中,基于子空间内分群信息量来实现高效的特征选择,从而提高细胞分群的准确性。此外,在ENCORE中提出了一种新的一致性聚类方法,以增强来自多个子空间的共有信号,同时保留各子空间的特有信号。与已有算法相比, ENCORE在大多数数据集上具有更优的分群性能、准确的标记识别以及更清晰的可视化效果。对于不同测序平台和归一化处理的数据,ENCORE的性能也更加稳定。ENCORE为单细胞数据的聚类、二维可视化分析提供了新的思路和方法,在细胞异质性研究和识别群体标记方面展现出巨大潜力。
撰稿人:林黎
校稿人:宋佳
原文链接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkaa1157/6030236
















 314
314











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









