







简介:X!Tandem作为生物信息学领域内用于蛋白质质谱数据分析的开源软件,采用C++编写,以其强大的数据处理能力在蛋白质组学领域得到广泛应用。它的开源特性提供了深入理解算法和定制开发的平台。本文档介绍了X!Tandem的工作流程,包括预处理、匹配肽段离子、评分与筛选以及后处理,并提供了特定版本的源码压缩包,供开发者和研究人员使用。源码中关键概念的理解,如质谱数据处理算法、搜索策略、评分系统、并行计算和可扩展性,对于提升生物信息学专业技能,创新科研技术具有重要意义。 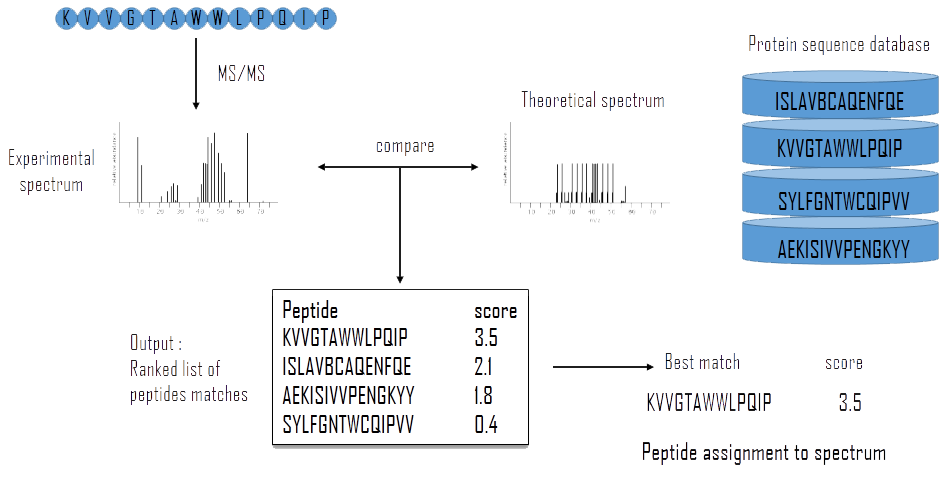
1. X!Tandem软件介绍
X!Tandem 是一款广泛应用于蛋白质组学领域的质谱数据分析软件。它以其高效的数据处理能力和灵活的算法配置而闻名,在科研和临床诊断中扮演着重要的角色。本章我们将简要概述 X!Tandem 的主要功能与特点,为进一步深入理解其在蛋白质研究中的应用奠定基础。
1.1 软件的主要功能
X!Tandem 的主要功能包括但不限于以下几点:
- 蛋白质鉴定 :通过质谱数据识别出样本中的蛋白质。
- 翻译后修饰(PTM)分析 :检测并定位蛋白上可能存在的修饰位点。
- 定量分析 :估算蛋白质相对丰度差异。
- 可视化工具 :提供直观的数据展示方式,如质谱图谱和蛋白质序列覆盖图。
1.2 应用场景与优势
X!Tandem 在多个科研场景中得到应用,包括但不限于:
- 蛋白质组学研究 :探索复杂生物样本的蛋白质组成。
- 临床研究 :分析病理样本,帮助诊断疾病。
- 药物研发 :分析药物作用的生物标记物。
相比其他同类软件,X!Tandem 的优势在于:
- 开源特性 :广泛的社区支持和持续的更新。
- 灵活的算法 :用户可根据研究需求定制算法参数。
- 跨平台兼容性 :支持多种操作系统,易于部署和使用。
在接下来的章节中,我们将深入探讨 X!Tandem 在蛋白质质谱数据分析中的应用,并详细解释其对生物信息学领域的贡献。
2. 蛋白质质谱数据分析应用
2.1 蛋白质质谱技术基础
2.1.1 质谱技术概述
质谱技术是现代生物化学和分子生物学中的一种重要技术,它通过测量样品中分子或分子片段的质量来分析物质的组成和结构。在蛋白质研究领域,质谱技术可以用于确定蛋白质的分子量、鉴定氨基酸序列、检测蛋白质修饰、测量蛋白质表达水平,以及进行蛋白质互作的分析等。
质谱技术的基本工作原理是将样品分子转换成带电的气相离子,然后通过电磁场分析其质量/电荷比(m/z)。通过分析不同m/z值离子的丰度,可以获得样品的质谱图。质谱仪由几个核心部分组成,包括离子源、质量分析器、检测器以及数据处理系统。每种类型的质谱仪在离子化方法、质量分析技术等方面有所差异,但它们共同目标是提供样品分子的精确质量和结构信息。
2.1.2 质谱数据的产生过程
质谱数据的产生是一个复杂的过程,它涉及到样品的制备、前处理、离子化、质量分析以及数据采集和处理等步骤。具体步骤通常包括:
- 样品制备:在进行质谱分析前,需要对生物样品进行适当的处理,以使蛋白质或肽段从复杂的生物基质中释放出来并转变为适合质谱分析的形式。
- 离子化:样品分子在离子源中被转化为气态离子,常用的方法包括电喷雾电离(ESI)和基质辅助激光解吸电离(MALDI)。
- 质量分析:通过不同类型的质谱仪,对形成的离子进行质量分析。常用的质量分析器有飞行时间(TOF)、四极杆、离子阱和傅里叶变换离子回旋共振(FT-ICR)等。
- 检测与记录:分析器中的离子被检测器捕获,并转换为电信号,然后被记录下来形成质谱图。
- 数据处理:最后,通过计算机软件对质谱图进行处理和分析,提取有用的信息,如分子量、肽段序列等。
2.2 质谱数据分析的重要性
2.2.1 数据分析在蛋白质研究中的角色
质谱数据分析在蛋白质研究中扮演着至关重要的角色。在蛋白质组学研究中,科学家通常需要从成百上千的样品中鉴定和定量蛋白质,而质谱技术就是实现这一目标的核心工具。数据分析不仅用于提取和验证肽段和蛋白质的身份信息,还用于评估蛋白质的相对和绝对表达水平、检测其翻译后修饰,以及探索蛋白质之间的相互作用网络。
通过对质谱数据的深入分析,研究人员可以:
- 映射出细胞或组织中的蛋白质组成。
- 理解不同生物状态下的蛋白质表达变化。
- 确定疾病的潜在生物标志物。
- 研究蛋白质功能和调控网络。
2.2.2 数据分析对生物信息学的贡献
生物信息学是一个跨学科领域,它结合了生物学、计算机科学、数学和统计学等多个学科的知识,用以解析生物大数据。质谱数据分析是生物信息学领域内的一项重要应用,它通过生物信息学的方法和技术,将质谱技术产生的大量原始数据转化为生物学上有意义的信息。
质谱数据分析对生物信息学的贡献主要体现在:
- 开发高效准确的算法来处理和解释质谱数据。
- 建立蛋白质组学数据库,实现数据的存储、检索和共享。
- 利用统计学方法对蛋白质组学数据进行定量分析和模式识别。
- 促进计算生物学模型的开发,用于模拟和预测蛋白质相互作用和代谢途径。
质谱数据分析的重要性在于其能够提供研究生物体结构和功能的基础数据,是生物信息学研究不可或缺的一部分。随着技术的发展和分析方法的改进,质谱数据分析将在未来生物学研究中扮演更加重要的角色。
3. C++编程语言特性
3.1 C++语言基础
3.1.1 C++语言的发展历程
C++,由Bjarne Stroustrup于1980年代初期开始开发,在其前身C语言的基础上引入了面向对象编程的特性。C++的第一个商业实现是1985年发布的,称为C with Classes。之后,随着1989年第一个完整的C++编译器发布,以及1998年和2003年的ISO标准的推出,C++逐渐发展成广泛使用的高级编程语言。
C++的设计初衷是提供一种能够使程序员更有效率的编程语言,同时仍然保持接近硬件的性能。这种特性使其成为系统软件、游戏开发、高性能服务器和客户端应用的首选语言。随着时间的推移,C++经历了多次重要的更新,包括模板、异常处理、STL(标准模板库)的引入,以及最新的C++20规范,增加了协程和概念等现代特性。
3.1.2 C++语言的核心概念
C++的核心概念覆盖了数据抽象、封装、继承和多态,这些是面向对象编程的基石。此外,C++还提供了对过程化和泛型编程的支持。
- 数据抽象 :它允许程序员通过创建抽象数据类型(ADT)来隐藏实现的复杂性,暴露给用户的是一个简单的接口。
- 封装 :把数据和操作数据的代码放在一起形成类,对类外隐藏内部实现细节。
- 继承 :使新的类可以从现有的类继承属性和方法,促进代码复用。
- 多态 :允许使用父类类型的指针或引用来引用子类的对象,实现接口的统一性。
C++还包含了模板和异常处理机制,提供了高度的泛型编程能力和错误处理能力。模板允许编写与数据类型无关的代码,异常处理机制使得错误处理变得更加模块化和集中。
3.2 C++在X!Tandem中的应用
3.2.1 X!Tandem对C++语言的需求分析
X!Tandem是一个复杂的生物信息学软件,用于分析蛋白质质谱数据。它对编程语言的要求很高,特别是在性能和资源管理方面。C++因其高性能和控制底层资源的能力成为首选。
- 性能 :质谱数据分析是一个计算密集型的过程,C++允许使用指针和内存管理优化来减少运行时间。
- 资源管理 :C++的栈和堆内存管理允许程序精确控制内存使用,这对于长时间运行的应用程序来说至关重要。
- 标准库支持 :C++标准模板库(STL)提供了强大的数据结构和算法,可以加速开发过程。
X!Tandem使用C++可以深入底层细节来优化算法性能,同时利用C++的面向对象特性来构建模块化和可维护的代码库。
3.2.2 C++特性的X!Tandem实现案例
在X!Tandem中,C++的特性被广泛应用于算法实现、数据结构设计和高效资源管理。
- 算法实现 :例如,在对质谱数据进行搜索匹配时,使用了高效的数据结构和搜索算法。
- 数据结构设计 :X!Tandem创建了复杂的类层次来表示蛋白质序列和相关的谱图数据。
- 高效资源管理 :在处理大规模数据集时,X!Tandem精心设计了内存分配策略来避免内存碎片。
为了展示C++在X!Tandem中的具体应用,下面是一个代码示例,说明了如何在C++中实现一个简单的蛋白质序列搜索算法:
#include <iostream>
#include <string>
#include <vector>
// 假设存在一个蛋白质序列类
class ProteinSequence {
public:
std::string sequence; // 存储蛋白质序列
// 其他成员函数和数据结构
};
// 简单的蛋白质序列搜索函数
std::vector<ProteinSequence> searchSequences(
const std::vector<ProteinSequence>& database,
const std::string& querySequence
) {
std::vector<ProteinSequence> result;
for (const auto& seq : database) {
if (seq.sequence.find(querySequence) != std::string::npos) {
result.push_back(seq);
}
}
return result;
}
int main() {
// 假设数据库和查询序列
std::vector<ProteinSequence> database = {/* ... */};
std::string querySequence = "ATCG";
// 执行搜索并获取结果
auto foundSequences = searchSequences(database, querySequence);
// 输出匹配的序列数量
std::cout << "Found " << foundSequences.size() << " matching sequences." << std::endl;
return 0;
}
在上述代码中,我们定义了一个 ProteinSequence 类来表示蛋白质序列,并实现了一个 searchSequences 函数,该函数遍历一个序列数据库,并搜索与查询序列匹配的所有序列。在主函数 main 中,我们创建了一个示例数据库和查询序列,然后调用搜索函数并输出匹配的序列数量。
请注意,实际的X!Tandem软件实现会更加复杂,涉及大量的蛋白质序列处理算法和优化技术,但上述代码提供了一个简化版本的实现,用以展示C++如何被应用于蛋白质序列数据的处理。
4. X!Tandem源码开源特性
4.1 开源软件的优势与挑战
4.1.1 开源软件的优势
开源软件,顾名思义,是其源代码对所有人公开,这种模式自从软件自由和开放源代码运动兴起以来,已经在IT行业获得了广泛的认可和应用。X!Tandem作为一款开源的蛋白质组学质谱分析软件,它具有以下几方面的优势:
首先,开源软件通常能够聚集一个由全球开发者构成的庞大社区,他们为软件的改进和维护提供无偿的支持。对于X!Tandem而言,这意味着可以快速地应对用户需求,增加新的功能,以及修正漏洞。
其次,透明度是开源软件的另一大优势。因为代码是公开的,所以任何人都可以检查软件的运行机制和算法实现,这大大增加了软件的可信度和可靠性。对于研究者来说,能够深入理解软件的内部工作机制是至关重要的。
再次,开源软件通常可以自由使用,不会产生高昂的许可费用。这对于预算有限的学术机构或小型企业来说是一个重要的优势。X!Tandem作为一个开源工具,为这些机构和企业提供了先进的蛋白质组学分析能力。
最后,开源软件的可定制性使得它能够更好地融入特定研究者的工作流程。用户可以根据自己的需要修改和扩展软件功能,而无需依赖于软件供应商。
4.1.2 开源软件面临的挑战
然而,尽管开源软件具有诸多优势,它也面临着一系列的挑战。其中最突出的是开源软件的支持和维护问题。由于缺乏商业盈利模式,开源项目可能会因为缺乏资金支持而难以持续。
此外,开源软件的开放性质也意味着任何人都可以使用和修改代码,这可能会导致代码质量和一致性的问题。没有严格的控制和管理,开源项目可能会产生分支版本,进而导致社区分裂,难以统一标准和功能。
开源社区的成员是志愿者,他们通常是基于兴趣而非义务参与项目。因此,社区活动可能会随着成员兴趣和可用时间的变化而波动,导致项目发展不稳定。
对于X!Tandem来说,这些挑战意味着开发团队需要更加注重社区的维护和用户的培训,以确保软件可以稳定和有效地服务于更广泛的用户群体。
4.2 X!Tandem的开源实践
4.2.1 X!Tandem的开源策略
X!Tandem的开源策略主要体现在其源代码的完全开放性以及开发过程的透明性。X!Tandem采用了GPL许可证,这要求所有基于X!Tandem开发的衍生软件也必须以相同的许可证发布,从而保证了软件生态的开放性。
为了进一步鼓励社区的参与和贡献,X!Tandem的开发团队建立了一套成熟的协作机制,包括公开的代码仓库、清晰的文档、定期的代码审查和维护。通过这些措施,开发团队确保了软件的持续进步和质量控制。
4.2.2 X!Tandem社区的贡献与维护
X!Tandem的社区是由广大用户和开发者共同构成的,他们通过多种渠道参与到软件的使用、测试、反馈和开发中。例如,社区成员可以通过邮件列表、论坛或GitHub上的Issue和Pull Request来提交问题和建议,以及贡献代码。
维护方面,X!Tandem项目拥有专门的维护团队,他们负责整合社区的贡献、更新软件和文档,以及管理社区活动。团队成员通过定期的视频会议和开发会议协调工作,确保项目的稳定运行和持续发展。
下面是一个简单的示例,展示了如何使用git命令与X!Tandem的GitHub仓库进行交互,获取源代码并创建自己的分支:
# 克隆X!Tandem的GitHub仓库到本地
git clone https://github.com/m迎ntandem/X!Tandem.git
# 进入源代码目录
cd X!Tandem
# 创建一个新的分支,用于自己的开发工作
git checkout -b new-feature-branch
在上述代码块中,用户首先克隆了X!Tandem项目的远程仓库到本地,然后切换到项目目录,并基于最新的主分支创建了一个新的分支,这个新分支可以用于开发新功能或修复bug等。这样的实践鼓励了社区成员参与到实际的开发过程中,共同推动X!Tandem的发展。
graph LR
A[开始克隆X!Tandem的GitHub仓库] --> B[进入源代码目录]
B --> C[创建新分支进行开发]
C --> D[提交代码到自己的分支]
D --> E[向X!Tandem的维护者发送Pull Request]
上图是一个简化的流程图,描述了开发者如何通过GitHub与X!Tandem项目进行交互。代码提交后,X!Tandem的维护者会审查代码,确认其符合项目标准后合并到主分支。通过这种机制,维护者保证了项目代码的稳定性和质量。
5. 质谱数据处理流程详解
5.1 质谱数据的预处理
5.1.1 数据清洗的方法与工具
质谱数据预处理的第一步通常是数据清洗。在这一阶段,我们致力于消除数据中的错误、异常值和不一致性,确保分析结果的准确性和可靠性。使用特定工具可以更有效地完成这一任务。
-
数据清洗工具介绍 在质谱数据处理领域,有几个流行的工具被广泛使用,如Skyline、OpenMS和ProteoWizard。它们具备以下功能:
-
去除噪声和背景信号 :利用特定算法,如Savitzky-Golay滤波器,去除质谱数据中的随机噪声。
- 识别并纠正数据偏差 :通过质量控制方法检测和校正仪器造成的偏差。
-
识别和填补缺失值 :缺失值可能因为仪器检测问题出现,利用统计方法进行填补。
-
代码实现
使用ProteoWizard中的 msconvert 工具来转换质谱数据格式,并进行初步清洗。以下是一个使用 msconvert 的命令示例:
bash msconvert --filter "peakPicking true 1-" -o output_directory input.mzML
参数解释: - --filter :启用过滤器, peakPicking true 指示进行峰识别, 1- 是指所有扫描。 - -o :输出目录,数据将被保存在指定的文件夹。 - input.mzML :输入的质谱数据文件(假设为mzML格式)。
通过这种方式,我们得到了预处理后的数据,为下一步的数据标准化和质量控制做好了准备。
5.1.2 数据标准化与质量控制
数据标准化和质量控制是质谱数据分析中的关键步骤,其目标是使数据在不同实验间具有可比性,并保证数据质量满足后续分析要求。
- 数据标准化方法 数据标准化可以采取以下几种方法:
- 校准曲线 :使用已知浓度的标准品制作校准曲线,将样本数据转换为真实浓度值。
- 内标校正 :在样本中加入一定量的已知浓度的内标物,通过内标物对样本数据进行校正。
-
归一化处理 :根据数据的总体分布,对数据进行归一化处理,使其在同一量级。
-
质量控制指标
在质量控制阶段,需要关注以下指标: - 信噪比(S/N) :用于衡量信号强度与背景噪声的比值。 - 重复性 :多次重复实验后数据的一致性。 - 分辨率 :仪器检测到相邻峰的分离程度。 - 质量误差范围 :测量质量与真实质量之间的差异。
- 代码示例
下面是一个使用R语言进行数据标准化的简单示例。假设我们有一个数据框 data ,其中包含质谱峰的强度值,我们可以使用以下代码来归一化这些值:
R # 安装和加载必要的包 if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("Affy") library(Affy) # 假设data是一个包含原始强度值的数据框 normalized_data <- normalizeBetweenArrays(data, method = "quantile")
这里使用了 normalizeBetweenArrays 函数,该函数属于 Affy 包,目的是在不同样本间进行标准化。参数 method = "quantile" 指定了使用分位数标准化方法。
5.2 质谱数据的分析与解释
5.2.1 蛋白质鉴定的算法流程
蛋白质鉴定是将质谱数据映射到特定蛋白质的过程。这一过程涉及多个步骤,包括数据匹配、数据库搜索、统计分析等。
- 算法流程
一个典型的蛋白质鉴定算法流程如下: - 谱图处理 :去除噪音、进行峰识别等预处理。 - 数据库搜索 :使用如X!Tandem、SEQUEST等软件工具将质谱数据与蛋白质数据库进行匹配。 - 评分和排名 :根据匹配的得分,将蛋白质进行排序,选出最佳匹配项。
- X!Tandem算法应用
X!Tandem使用一种概率评分算法,它基于质谱数据与理论谱图的匹配程度。它的优势在于能够处理大规模数据集,并对蛋白质变异体进行较好的识别。
- 代码示例
下面是一个使用X!Tandem进行蛋白质鉴定的基本代码示例。假设已安装并配置好X!Tandem:
bash tandem -f input.mgf -d database.fasta -o output.xml
参数说明: - -f input.mgf :输入文件,假设为MGF格式的质谱数据。 - -d database.fasta :参考蛋白质数据库,假设为FASTA格式。 - -o output.xml :输出结果,保存为XML格式。
5.2.2 蛋白表达量的评估方法
在蛋白质鉴定之后,我们常常需要评估蛋白质的表达水平。这里通常使用的方法包括基于质谱强度的方法和基于标签的方法。
- 基于质谱强度的方法 质谱强度指的是蛋白质特征峰的强度,可以通过以下公式计算表达量:
- iTRAQ/TMT标签 :标记不同样本,允许在同一个质谱运行中比较不同样本。
-
无标签定量 :直接比较不同实验条件下相同蛋白质的峰强度。
-
代码示例
以下使用R语言中的 MSnbase 包来评估表达量。假设 expression_data 包含了质谱数据:
R library(MSnbase) # 读取质谱数据 raw_data <- readMSData(expression_data) # 标准化质谱数据 norm_data <- normalize(raw_data, method="quantiles") # 估算表达量 expression_values <- esetApply(norm_data, median, na.rm=TRUE)
在这个示例中,我们首先加载 MSnbase 包,然后读取质谱数据,接着使用标准化方法对数据进行标准化处理,并最终计算出表达量。
总结
在本章中,我们深入探讨了质谱数据处理的详细流程。从数据预处理到分析和解释,每一步都是为了获得可靠且具有生物学意义的结果。数据清洗与标准化是质谱数据分析的基础,确保了数据的质量。随后,使用高级算法和工具进行蛋白质鉴定,并通过定量方法评估蛋白表达量。整个流程的每一步都要求操作人员具有深厚的专业知识,并熟悉相关的工具和技术。
本章的内容通过实际的代码示例和工具应用,为读者提供了一个清晰的质谱数据处理的实践框架。这不仅有助于理解质谱数据的处理流程,也为之后章节中对X!Tandem软件的深入学习打下了坚实的基础。
6. 特定版本源码压缩包介绍及源码关键概念深入理解
在生物信息学研究和软件开发中,理解特定版本的源码压缩包和源码关键概念是至关重要的。本章我们将深入探讨源码压缩包的结构及其包含的文件,以及在源码层面上如何理解X!Tandem的功能实现。
6.1 版本控制与源码管理
6.1.1 版本控制系统的选用
在生物信息学领域,选择合适的版本控制系统对协作和代码管理至关重要。目前广泛使用的是Git,因为它提供强大的版本控制功能,支持分布式开发模式,非常适合于分散在世界各地的研究团队协作开发。
例如,X!Tandem项目团队采用Git作为其源码版本控制系统。它允许开发者在本地进行更改,然后通过网络将更改推送到远程仓库或从中拉取更改。
# 在本地仓库中初始化一个新的Git仓库
git init
# 将更改添加到仓库
git add .
# 提交更改到本地仓库
git commit -m "Initial commit of X!Tandem source code"
# 将本地仓库内容推送到远程仓库
git push origin main
6.1.2 源码管理策略与实践
源码管理不仅仅是备份和版本控制,它还包括代码审查、分支管理等策略。例如,X!Tandem团队使用主分支(main)来保存稳定版本的代码,而开发分支(develop)则用于集成新功能。
团队成员在开发新功能时会创建自己的功能分支(feature branch),在完成后通过Pull Requests(PR)请求将代码合并到develop分支。
graph LR
A[main branch] -->|Merge| B(develop branch)
B -->|Merge| C(feature branches)
C -->|Pull Request| B
6.2 源码关键概念深入理解
6.2.1 X!Tandem源码结构解析
X!Tandem的源码结构清晰地分为了多个模块,每个模块负责软件的不同部分。例如,核心算法模块负责蛋白质鉴定,而用户界面模块则负责与用户交互。
源码通常包含以下关键文件和目录结构:
- core/ : 包含核心算法和数据结构定义。
- interface/ : 包含用户界面的实现代码。
- util/ : 包含各种工具函数和辅助模块。
通过查看 README.md 文件,可以了解每个模块的详细功能和如何编译运行整个项目。
6.2.2 源码中关键模块与功能的实现
要深入理解X!Tandem的源码,我们首先需要熟悉其核心模块如何实现关键功能。以蛋白质鉴定模块为例,它通常包含以下几个关键组件:
- Spectrum Matching : 实现质谱图与理论谱图的匹配算法。
- Database Search : 使用数据库进行蛋白质序列搜索。
- Scoring System : 算法评分系统,评估匹配质量。
例如,下面是一个简化的伪代码片段,展示了如何在X!Tandem中使用一个基本的评分函数进行谱图匹配:
class SpectrumMatch {
public:
double score; // 匹配评分
// 其他成员变量和方法...
// 计算匹配评分的函数
double calculateScore() {
// 根据匹配的质量计算评分
score = computeQualityOfMatch();
return score;
}
private:
double computeQualityOfMatch() {
// 具体计算匹配质量的逻辑...
}
};
// 使用SpectrumMatch进行匹配和评分
SpectrumMatch match;
match.calculateScore();
// 输出评分结果
std::cout << "Match score: " << match.score << std::endl;
通过学习源码中的这些关键概念,我们可以更好地理解X!Tandem的工作原理,并且有能力对其进行优化和贡献。
在下一章中,我们将继续深入探讨质谱数据处理流程的详解,以及如何将理论应用到实践中,增强对X!Tandem软件的全面理解和应用能力。
简介:X!Tandem作为生物信息学领域内用于蛋白质质谱数据分析的开源软件,采用C++编写,以其强大的数据处理能力在蛋白质组学领域得到广泛应用。它的开源特性提供了深入理解算法和定制开发的平台。本文档介绍了X!Tandem的工作流程,包括预处理、匹配肽段离子、评分与筛选以及后处理,并提供了特定版本的源码压缩包,供开发者和研究人员使用。源码中关键概念的理解,如质谱数据处理算法、搜索策略、评分系统、并行计算和可扩展性,对于提升生物信息学专业技能,创新科研技术具有重要意义。

















 998
998

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









