






论文链接:https://academic.oup.com/bioinformatics/article-abstract/38/9/2452/6543608
DOI:https://doi.org/10.1093/bioinformatics/btac138
期刊:Bioinformatics
代码链接:https://github.com/ChunhuaLiLab/SREPRHot
发布时间:07 March 2022
前言
识别蛋白质-RNA相互作用中的结合位点对于理解其潜在的识别机制和药物设计至关重要。这些实验方法有许多局限性,因为它们通常是耗时耗力的。因此,迫切需要开发一种有效有效的方法。
在这里,作者提出了SREPRHot,一种预测位点的方法,定义为丙氨酸突变产生结合自由能变化2.0千卡/摩尔的残基。为了解决数据集的不平衡问题,利用合成少数过采样技术(SMOTE)生成少数样本,以实现数据集的平衡。此外,除了传统的特征外,还使用了两种新的特征,即作者之前开发的残差界面倾向和使用节点加权网络获得的拓扑特征,并提出了一种有效的随机分组特征选择策略,结合两步方法来确定最优特征集。最后,采用堆叠集成分类器建立了该模型。结果表明,SREPRHot在独立测试数据集上的SEN、MCC和AUC分别为0.900、0.557和0.829。对比研究表明,SREPRHot具有较好的应用性能。
一、Introduction
蛋白质-RNA相互作用通过调节基因表达过程的不同步骤,在各种生物过程中发挥着关键作用。相互作用的异常可能导致多种疾病,如癌症和神经系统疾病。众所周知,有一小部分界面残基,被称为结合位点,贡献了目标RNA的大部分结合自由能。因此,蛋白质-RNA相互作用中的可靠位点识别对于理解其潜在的识别机制和设计药物至关重要。在实验中,通过评估其突变为丙氨酸后的结合自由能变化(DDG),可以找到一个位点残基。
到目前为止,由于现有的实验数据有限,预测蛋白质-RNA相互作用位点的方法还很少,这滞后于蛋白质-蛋白质位点的预测。
(1)2016年,Barik等人提出了一种随机森林(RingHotSPRF)模型,该模型利用界面残基的结构和物理化学特征来预测RNA结合残基突变的DDG范围。
(2)2018年,Pan等人开发了PrabHot,这是一种性能更好的工具。
(3)2019年晚些时候,Deng等人引入XGBPRH方法,采用McTWO算法选择6个最优特征训练极限梯度增强(XGBoost)分类器,MCC提高到0.661。
除了分类器外,用于位点预测的特征也很重要。现有的方法主要使用一些基于序列和结构的特征。事实上,我们还需要探索其他特征来改进蛋白质-RNA结合热点的预测。在之前的工作中,作者从蛋白质-RNA相互作用中提取了残基-核苷酸成对倾向潜力,在蛋白质-RNA相互作用预测和界面残基识别方面表现良好。
此外,对于氨基酸网络(AAN)模型中的残基拓扑特征,已成功用于探索功能位点,包括催化、变构和配体结合残基。通常,拓扑特征由传统的非加权AAN模型获得,该模型忽略了残基节点的异质性,而该异质性对于区分结构或功能上重要的残基至关重要。鉴于此,已经开发了许多加权AAN模型,其中Yan等人开发的节点加权网络能够表征节点异质性,并广泛应用于功能残差预测。
对于一个相对较小的样本量,选择一个重要特征的子集对于建立一个有效的预测器是很重要的。常用的特征选择方法包括最小冗余最大相关性(mRMR;Peng et al.,2005)、RF(Breiman,2001)和Boruta,它们的性能在小样本量下不是很理想。作者解决这一问题的策略是,首先将样本分成几个子集,分别对所有子集进行特征选择,然后保留通常选择的特征作为最优特征集。研究结果证明了该策略的有效性,下面我们称之为随机分组特征选择策略。
通常采用过采样和欠采样技术对不平衡数据进行预处理,其中合成少数过采样技术(SMOTE)经常应用于数据挖掘领域。与通过简单随机复制生成少数类样本的朴素随机过采样算法不同,SMOTE方法通过特征空间中的某些操作生成合成样本,在一定程度上避免了过拟合问题。最近的几项研究已经成功地利用SMOTE有效地改进了蛋白质-蛋白质相互作用位点和药物-靶点相互作用的预测。
SREPRHot引入了SMOTE来平衡数据类。通过随机分组特征选择策略,结合从蛋白质序列和结构中提取的8种候选特征,包括残基-核苷酸成对倾向势和残基拓扑特征,选择了最优的特征子集。然后利用这些特征训练一个堆叠集成分类器来构建位点预测器。SREPRHot方法的框架如图1所示。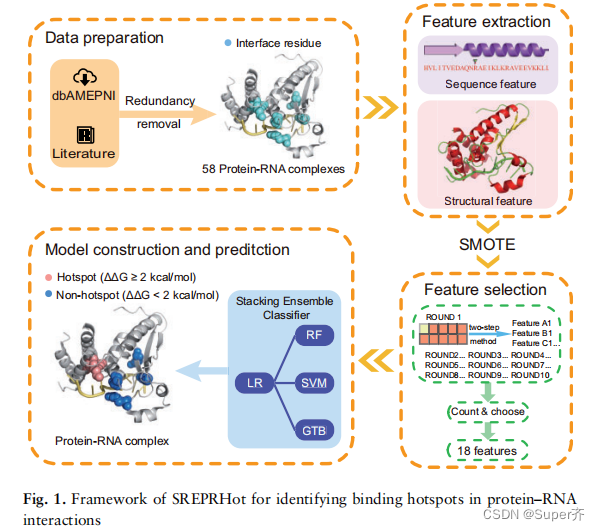
二、Materials and methods
2.1 Training and testing datasets
作者总共从81个复合物中收集了334个残基突变。为了消除冗余,使用CD-HIT排除了序列相似度>40%的蛋白质。在此之后,只保留了与目标RNA后的界面残基。最后获得了58个复合物中的229个残基突变。其中,将PrabHot和XGBPRH方法作为测试数据集的15个复合物作为独立测试数据集,其余的作为训练数据集。
与现有的以DDG为1.0千卡/mol的界面残基为热点的方法不同,我们的方法采用了DDG为2.0千卡/mol的准则。因此,在训练数据集中有35个正样本和136个负样本。
2.2 Feature extraction
从8种类型中提取了120个特征。关于这些特性的更多细节将在下面进行描述。
2.2.1 Physicochemical characteristics of amino acids
10种氨基酸的理化性质取自AAIndex数据库和文献,包括原子数、静电电荷数、潜在氢键数、疏水性、亲水性等等。
2.2.2 Secondary structural features
应用SPIDER3计算蛋白质的二级结构特征,包括主链扭转角(u和w)、Ca原子之间的主链角(h和s)以及三种二级结构的概率:-螺旋、-链和随机螺旋。
2.2.3 Depth index and protrusion index
结合界面上的几何形状的互补性对蛋白质-rna的相互作用很重要。深度指数(DPX)和突出指数(CX)分别被用来表征被其他非氢原子包围的原子的嵌入和突出条件。我们使用PSAIA计算指数的蛋白质在绑定和非绑定状态包括所有原子的DPXs和cx的标准差,和侧链原子的DPXs和cx的标准差。此外,还计算了束缚态和非束缚态之间所有原子和侧链原子之间的均值和标准差。
2.2.4 Solvent accessible surface area
利用Naccess计算了复合物和单体中残留物的溶剂可及表面积(SASAs),共包含10个属性:所有原子、总侧链原子、主链原子、非极性原子和所有极性原子的绝对值和相对值。此外,还计算了它们的变化及其对应的平方根。
2.2.5 Position-specific scoring matrix
位置特异性评分矩阵(PSSM)给出了每种氨基酸残基在每个位置出现的概率,这反映了一个残基位置的进化信息。对于具有N个残基的蛋白质,其PSSM矩阵的大小为N*20,每一行封装了一个残基位置的进化信息。通过PSI-BLAST搜索NCBI非冗余蛋白质序列数据库,计算出蛋白质的PSSM。
2.2.6 Solvent exposure features
半球暴露(Half-sphere exposure)是一种描述残基和溶剂分子之间接触的溶剂暴露措施,已被证明对蛋白质结构和功能预测很重要。HSE是一个二维度量,其中残差的空间球被分为两部分:HSE-up(残差侧链方向上的上球)和HSE-down(相反方向上的下球)。采用HSEpred计算溶剂暴露特征HSE-up和HSE-down,并计算残留接触数(CN)。
2.2.7 Residue interface propensity
残基界面倾向(IP)来自我们之前获得的20*4残基-核苷酸成对倾向潜力,来自251个蛋白质-rna相互作用,后来更为694个相互作用。一个残基-核苷酸对的倾向是由它所观察到的概率除以它在界面上发生的预期概率得到的。在这里,一个残基类型的IP被表示为它对四种核苷酸的配对倾向的平均值。
2.2.8 Residue topological features from AAN
与传统的非加权AAN相比,考虑了残差异质性的节点加权AAN可以更好地反映残差的拓扑性质。本文除未加权AAN外,分别构建了基于残差质量、疏水性、极性和溶剂可及性的四个节点加权AAN,并利用R包‘NACEN”计算了相应的残差拓扑特征,包括度、中间度中心度和接近中心性。
2.3 SMOTE dataset balancing algorithm

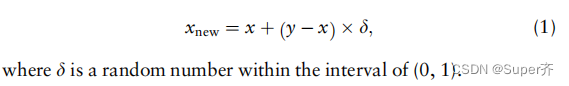
2.4 Feature selection
在此,提出了一种新的随机分组策略,并结合了一种两步算法来选择最优的特征子集。首先,将训练数据集随机分为10个等量的组,进行10轮特征选择,每轮使用10个组中的9组。然后记录每一轮中选择的特征,最终只保留10轮中选择的不少于2次的特征作为最优特征集。
每一轮都采用两步法。首先,结合了mRMR和决策树(DT)方法来对候选特征的重要性进行排序。然后,使用序列正向选择结合默认参数的支持向量机(SVM),通过重复10次交叉验证最大化Ec分数,从重要性列表中前60名中确定最优特征组合。Ec分数的计算方法为
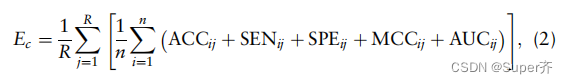
式中,n和R(采用10和5)分别为交叉验证折叠数和n倍交叉验证的次数,ACC、SEN、SPE、MCC和AUC分别为准确性、敏感性、特异性、Matthew相关系数和AUC评分。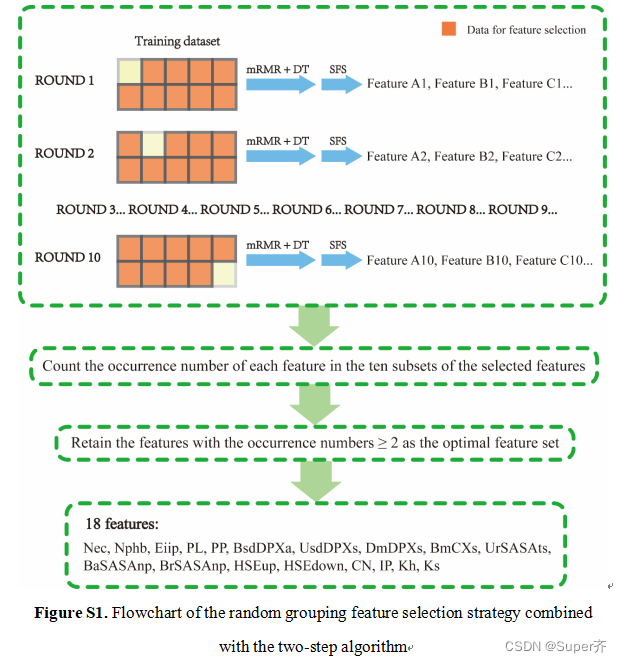
2.5 Stacking ensemble classifier
堆叠是一种通过元分类器结合多个基分类器的集成学习策略,已被许多研究证明比单一分类器性能更好。在这里,我们应用三种增强分类器梯度树增强,RF和SVM作为基础分类器,逻辑回归作为元分类器。
2.6 Performance evaluation
SREPRHot通过10倍交叉验证对训练数据集进行调优,并在独立的测试集上进行测试。评价指标包括准确性(ACC)、敏感性(SEN)、特异性采用(SPE)、精度(PRE)、F1评分(F1)和MCC,其定义如下:
三、Results
3.1 Advantage of the SMOTE algorithm
使用热点值DDG为2.0千卡/mol的准则导致正负样本之间的高度不平衡,使得特征选择和模型构建主要以负样本为主,不利于模型构建。采用SMOTE算法在训练集中生成少数(正)类样本来平衡数据。为了探索是否样本比例1:1可以提高模型性能,我们比较结果得到的模型训练平衡数据打击和随机重复过采样技术(简单的复制操作),和初始不平衡的,如表1所示。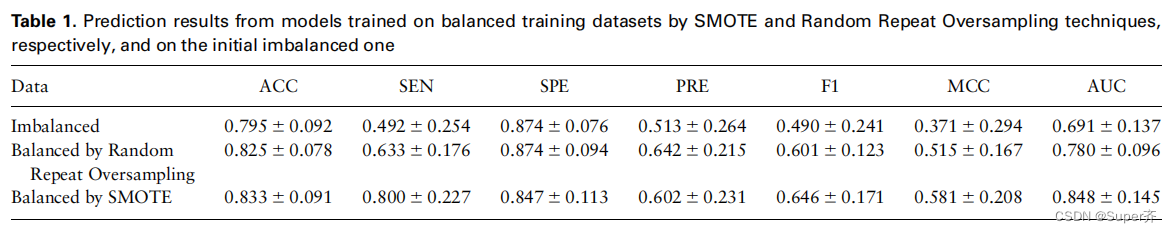
从表1中可以看出,与在不平衡数据上训练的模型的结果相比,在不平衡数据上训练的模型的相应结果有了明显的改善。此外,在SMOTE处理的数据集上训练的模型比随机重复过采样处理的模型预测效果更好,SEN、MCC和AUC分别提高了26.4%、12.8%和8.7%。我们认为,这种改进的原因是,随机重复过采样技术生成的样本只是原始正样本的副本,而没有增加任何新信息,这可能会导致一定程度的过拟合。
3.2 Evaluation of different feature selection methods
为了探究随机分组策略和新算法的优势,比较了四种经典方法mRMR、RF、Boruta和SFS的性能,以及我们的随机分组策略结合两步方法的性能。结果如表2所示。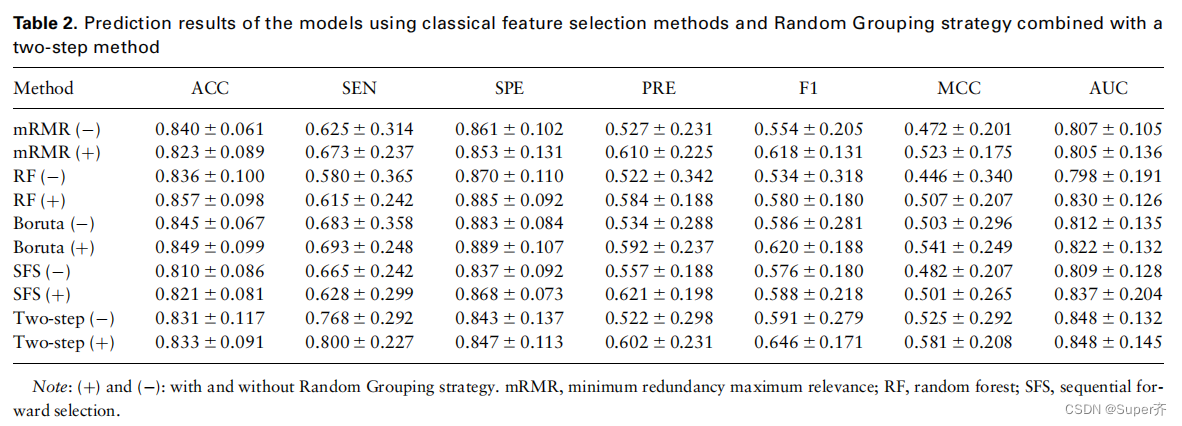
如表2所示,在没有随机分组策略的特征选择方法中,两步法的性能最好。此外,考虑到该策略,每种方法的性能都有一定程度的提高,特别是在SEN、F1和MCC分数方面。因此,我们提出的随机分组策略结合了一个两步算法,为我们的模型选择最优特征,其表现明显优于其他方法。我们推测可能的原因是两步法考虑了特征之间的互补性,减少了过拟合,随机分组策略在一定程度上减少了离群样本对特征选择的影响。
通过随机分组策略结合两步算法进行降维后,我们最终得到了18个特征的最优集,如补充表S5所示。在18个特征中,9个特征是基于序列的四种类型(氨基酸的理化特征、PSSM、溶剂暴露特征和IP),其他9个特征是基于其他3种类型(DPX和CX、SASA和拓扑特征)。需要指出的是,我们选择了我们提出的残基IP和节点加权AAN中的两个拓扑特征作为最优特征,据我们所知,这是首次用于蛋白质-rna热点预测。IP表示一种氨基酸出现在界面上的倾向,而热点则是一种特殊的结合位点,我们认为这可能是IP有助于预测结合界面上的热点的原因。在拓扑特征方面,一些研究证明,考虑网络中节点的异质性有助于功能残差的识别。
3.3 Comparison between different machine learning methods
我们需要选择一个合适的机器学习方法来建立我们的模型。为此,我们在训练数据集上使用10倍交叉验证,比较了6个经典分类器的性能,结果如补充表S6所示。与分类器kNN、自适应增强和极限梯度增强(XGBoost)相比,GTB、RF和SVM在PRE、F1和MCC分数方面表现最好。鉴于此,我们采用GTB、RF和SVM三个分类器作为我们SEC的第一层分类器,LR作为第二层输出最终结果,这在一定程度上降低了过拟合的风险。结果表明,SEC的性能一般远远优于其他分类器,ACC、PRE、F1、MCC和AUC分别为0.833、0.602、0.646、0.581和0.848。因此,SEC由于其优越的性能而被用作SREPRHot的机器学习分类器。
3.4 Performance comparison of SREPRHot with other approaches
为了精确估计SREPRHot,在训练数据集上重复了10倍交叉验证,得到ACC、SEN、F1、MCC和AUC值分别为0.818 6 0.016、0.814 6 0.036、0.638 6 0.022、0.565 6 0.023和0.859 6 0.019。结果表明,该模型的性能是相对稳定和稳健的。
此外,还比较了SREPRHot在独立测试数据集上的性能与现有方法PrabHot、XGBPRH和HotSPRing的性能,结果如表3所示。

需要指出的是,前两种方法是用于预测DDG阈值为1.0千卡/mol的热点地区,后者用于预测一个残基突变的DDG范围。XGBPRH的开发者Deng等人为了比较XGBPRH和HotSPRing的性能,采用了1.0千卡/摩尔的阈值。表3中与HotSPRing对应的结果来自文献,因为HotSPRing目前还不可用。从表3中可以看出,XGBPRH的性能一般最好,SEN、MCC和AUC分别为0.909、0.661和0.868。考虑到我们的方法采用了更严格的DDG 2.0 kcal/mol的标准,SREPRHot取得了良好的性能,SEN、MCC和AUC分别达到0.900、0.557和0.829。比较表明,我们的方法显示出了很好的性能,可以作为使用1.0千卡/摩尔的方法的补充。
Conclusion
有效预测蛋白质-rna相互作用中的结合热点对于理解其特定的识别和相互作用机制至关重要。本文提出了一种新的识别结合热点的SREPRHot识别方法,该方法以预测蛋白残基的18个特征作为输入,并给出其分类结果作为输出。为了解决采用更严格的热点标准DDG为2.0千卡/摩尔而不是1.0千卡/摩尔导致的数据类不平衡问题,采用SMOTE算法生成少数(正)类样本,达到数据类平衡。除了传统的序列特征和结构特征外,还提取了两种新的特征类型,即我们开发的残差IP和节点加权AAN中的拓扑特征作为候选特征。从中,我们提出的随机分组特征选择策略结合两步法,挑选出一个最优特征集。最后,采用叠加集成模型通过LR组合三个性能良好的分类器GTB、RF和SVM来构建分类方法。与现有的方法相比,SREPRHot取得了较好的性能。我们认为,我们的方法是预测绑定热点的一个新的开始,并且所提出的对数据进行预处理和选择最优特征的策略也可以作为未来预测工作的参考。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









