




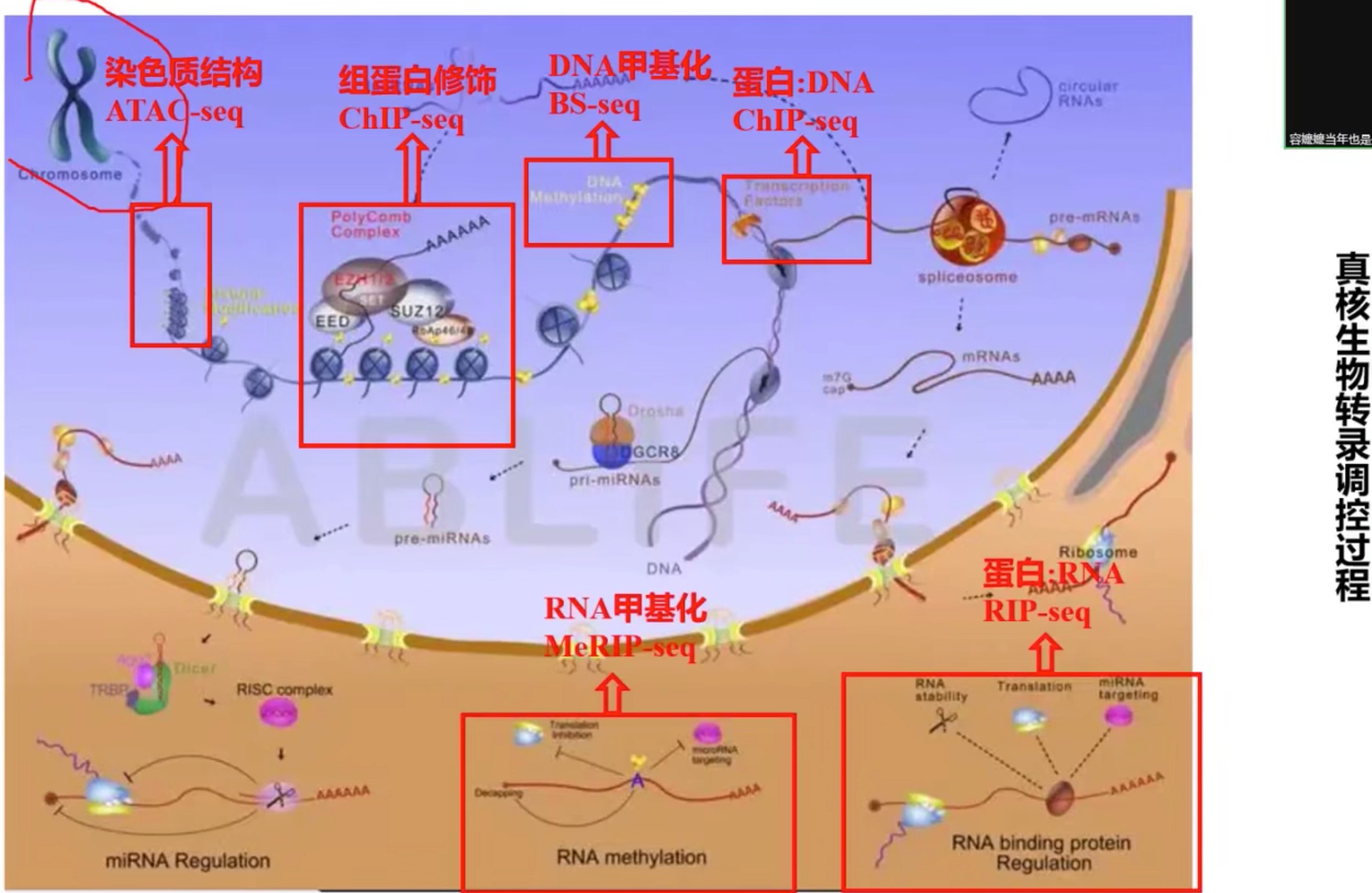
 ManolisKellis教授的课程介绍了基因调控的基础,包括DNA甲基化、组蛋白修饰的作用机制和功能,以及关键的实验技术如CHIP-seq、DNase-seq和ATAC-seq,这些技术用于揭示基因表达调控的细节。
ManolisKellis教授的课程介绍了基因调控的基础,包括DNA甲基化、组蛋白修饰的作用机制和功能,以及关键的实验技术如CHIP-seq、DNase-seq和ATAC-seq,这些技术用于揭示基因表达调控的细节。
文章目录
来自Manolis Kellis教授(MIT计算生物学主任)的课
油管链接:Regulatory Genomics - Deep Learning in Life Sciences - Lecture 07 (Spring 2021)
本节课分为三个部分,本篇笔记是第一部分。
本节探讨基因调控的生物学基础、DNA motifs以及用于探测基因调控的关键技术(CHIP-seq、DNase-seq以及ATAC-seq)。主要是为后面的内容打下基础,为深度学习所用的数据进行说明
- 课程大纲

Biological foundations of gene regulation
-
DNA甲基化:
- 原因: DNA甲基化是直接作用于DNA分子的一种修饰。当某些胞嘧啶(C)位点被添加上一个甲基团时,它们就被称为被甲基化了。
- 机制: DNA甲基化通常会导致基因的沉默,因为这种修饰阻止了转录因子和其他蛋白质与DNA结合。此外,甲基化的DNA会吸引某些蛋白质(如甲基化DNA结合蛋白),这些蛋白质可以进一步改变染色质的结构,使其更为紧凑。
- 功能: DNA甲基化在许多生物过程中起到关键作用,如基因沉默、X染色体失活、创伤应激反应和癌症。
-
组蛋白修饰:
- 原因: 组蛋白修饰是在组蛋白的特定氨基酸残基上发生的化学变化。例如,赖氨酸上可以添加甲基、乙酰或磷酸化团等。
- 机制: 组蛋白修饰可以改变染色质的紧凑度,从而影响DNA的可及性。例如,某些组蛋白修饰可以使染色质更为放松,促进基因的表达,而其他修饰则导致染色质更为紧凑,抑制基因的表达。
- 功能: 组蛋白修饰在基因表达调控、DNA损伤修复、细胞周期调控等多种过程中都发挥着重要作用。
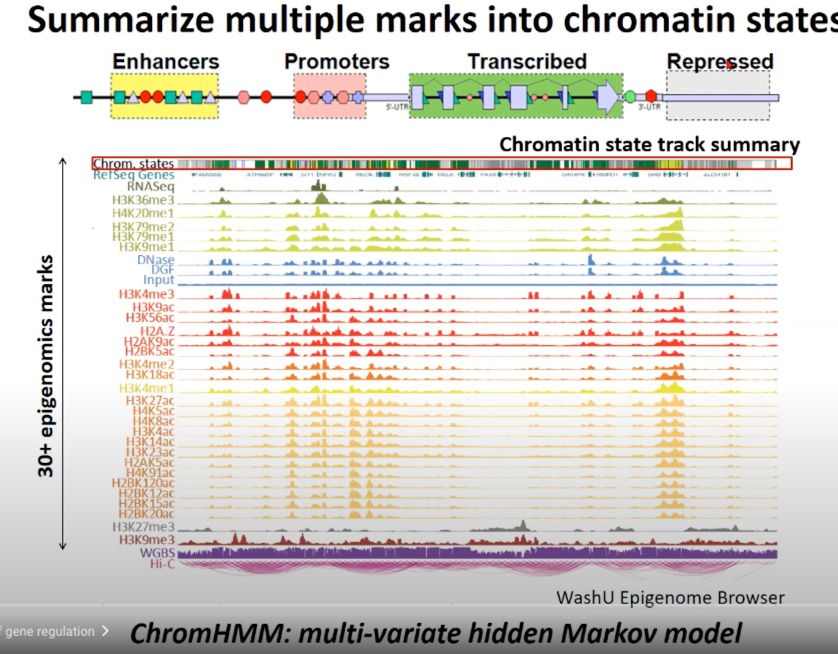
在使用ChromHMM或类似的方法进行染色质状态分析时,染色质状态被视为隐藏状态,而实际的表观遗传标记(例如组蛋白修饰)被视为观察到的数据。
隐马尔可夫模型(HMM)的工作方式是,基于观察到的数据(在这里是各种表观遗传标记)来推断最有可能的隐藏状态序列(在这里是染色质状态)。因为染色质状态本身不能直接测量,所以需要通过这种统计方法来推断它。
例如,如果一个基因区域有H3K4me3和H3K27ac的组蛋白修饰,但没有H3K27me3,那么HMM可能会推断这个区域是一个活跃的启动子。这种推断是基于已知的关于这些修饰与基因活性之间的关系。
DNA motifs
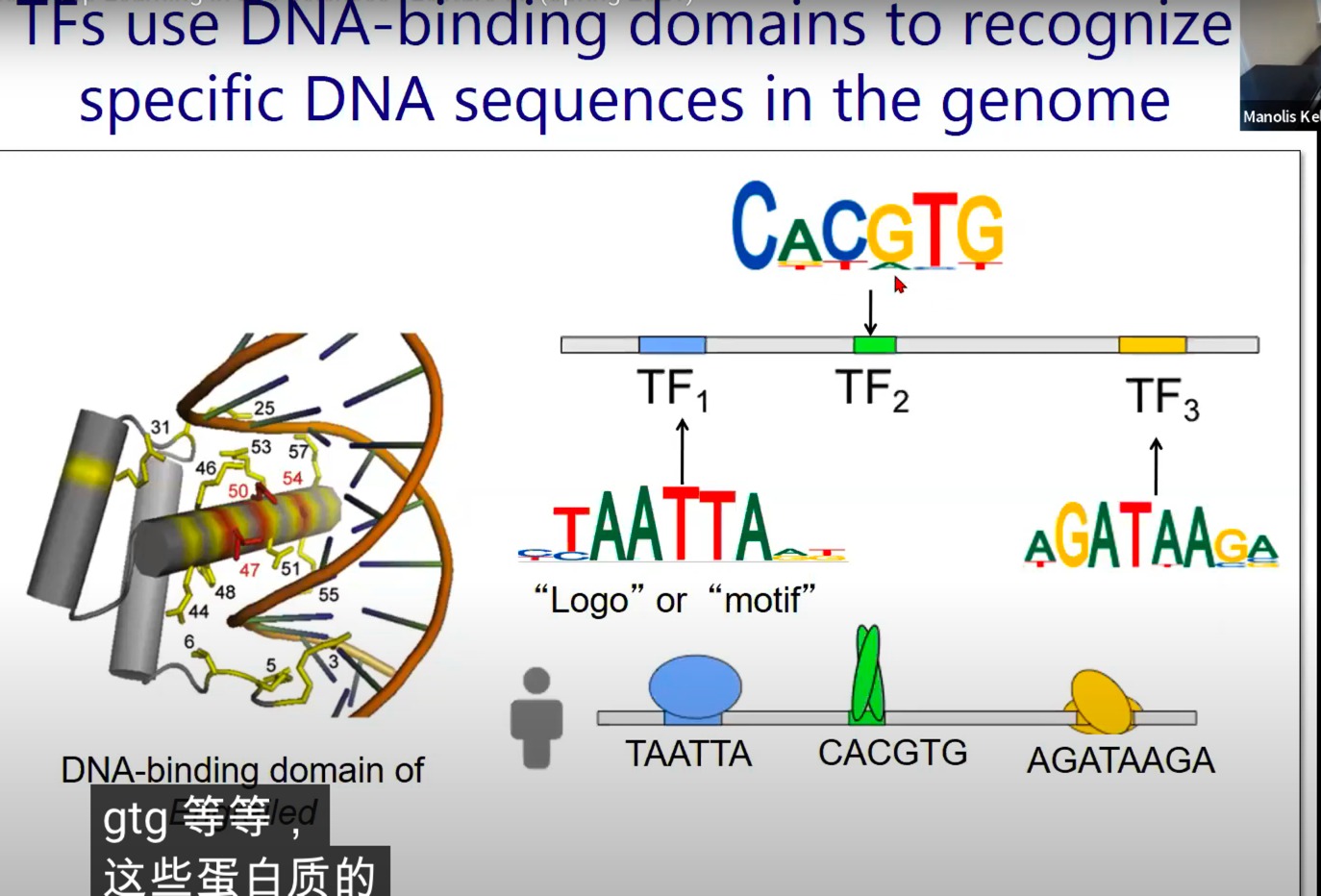
- DNA结合结构域:这是转录因子中的一个区域,能够特异性地与DNA中的特定序列结合。
- 识别特定序列:展示了三种不同的转录因子(TF1、TF2和TF3)如何识别和结合到特定的DNA序列上。例如,TF1与TAATTA序列结合,TF2与CACGTG结合,而TF3与AGATAAGA结合。
-
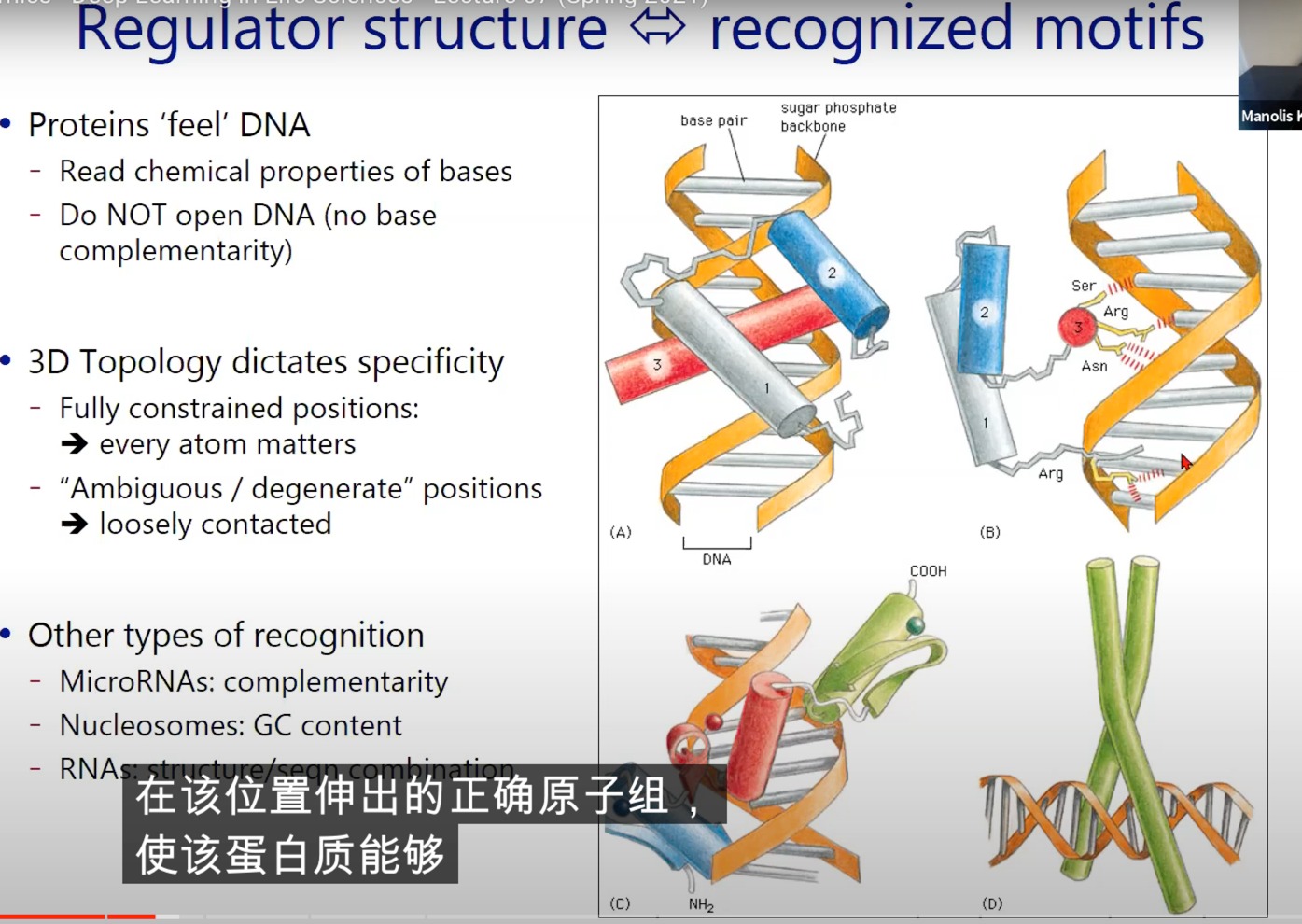
蛋白质“感觉”DNA:
- 蛋白质可以“读取”DNA碱基的化学特性。
- 蛋白质并不打开DNA(不需要碱基互补性来与DNA结合)。
-
3D拓扑结构决定特异性:
- 完全受限位置:在这里,每一个原子的位置都很关键。这意味着蛋白质与DNA的结合是非常精确的,每一个原子的位置都有其特定的功能。
- “模糊/退化”位置:这些位置的接触是松散的,不需要完全匹配。这意味着在这些位置,蛋白质与DNA的结合有一定的灵活性。
-
其他类型的识别:
- MicroRNAs:这是一种小的非编码RNA,其识别原理是基于碱基的互补性。
- 核小体:它们的识别与GC含量有关。GC含量是指DNA序列中鸟嘌呤(G)和胞嘧啶(C)的百分比。某些核小体更倾向于与GC含量较高的区域结合。
- RNA结构和组合:这里提到RNA的结构和组合也可以影响与其他分子的识别和结合。
Technologies for probing gene regulation
CHIP-seq
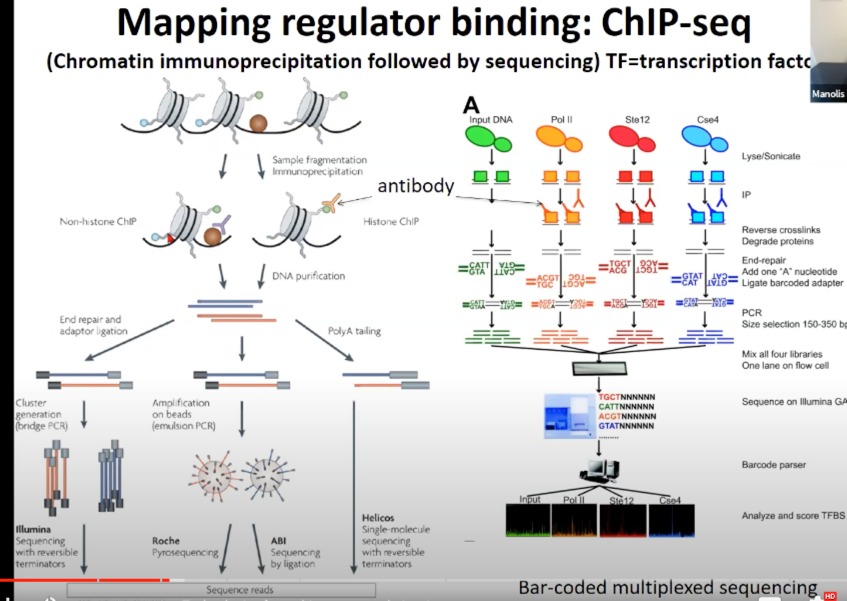
ChIP-seq(染色质免疫沉淀后续测序),这是一种用于确定DNA中某特定蛋白质(例如转录因子或组蛋白修饰)绑定位置的技术。
-
样本制备:首先,细胞内的DNA和与其结合的蛋白质通过形式化物固定,这样可以保持其相互作用。
-
Lysing/Sonication:细胞裂解,随后通过超声波破碎DNA,形成较小的片段。
-
Immunoprecipitation (IP):使用特定的抗体来沉淀目标蛋白质及其结合的DNA。这里展示了与非组蛋白结合(Non-histone ChIP)和与组蛋白结合(Histone ChIP)的情况。
-
DNA提纯:将绑定的DNA从蛋白质中分离出来。
-
End Repair & Adaptor Ligation:修复DNA的末端并连接一个适配器,以便于下一步的扩增。
-
PolyA tailing:添加PolyA尾,有助于下一步的测序。
-
扩增和测序:
- Amplification on beads (emulsion PCR):在微珠上进行扩增,使得每个珠子上只有一个DNA模板。
- Cluster generation (bridge PCR):在Illumina测序平台上,形成簇或集群,每个簇由同一个模板扩增的多个DNA片段组成。
- 下面部分展示了不同的测序技术,包括Illumina、Roche Pyrosequencing、ABI以及Helicos等。
-
Bar-coded multiplexed sequencing:使用带有不同条形码的适配器,使得多个样本可以在同一个通道上进行测序,之后再通过条形码进行区分。
-
数据分析:最后,得到的测序读数被解析并分析,以确定目标蛋白质在基因组中的结合位置。图中展示了四种不同的蛋白质(Input, Pol II, Ste12, Cse4)的结合位置,这可以帮助研究者了解这些蛋白质如何调控基因的表达。
DNase-seq
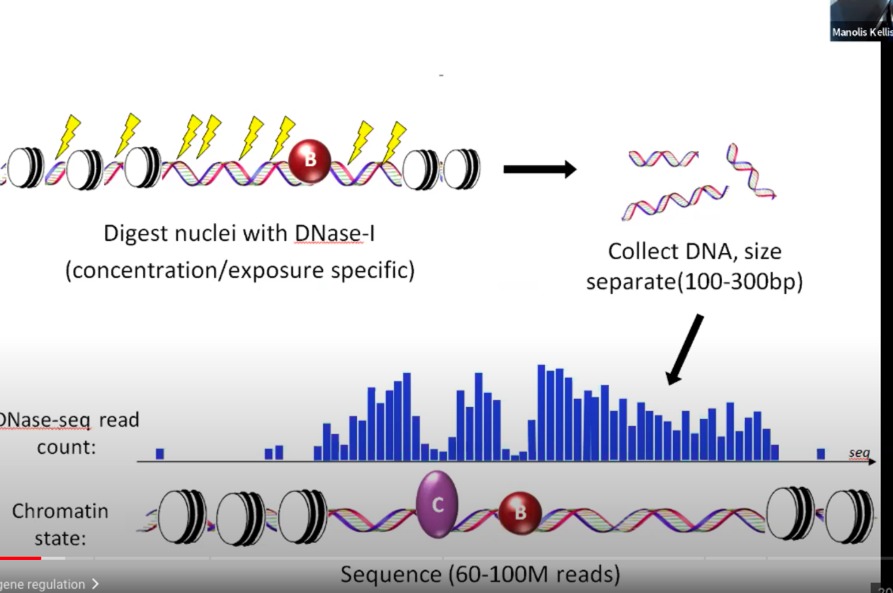
DNase-seq是一种用于鉴定基因组中开放染色质区域的技术。开放染色质区域通常与基因的转录活性相关,并可能包含转录因子结合位点。
以下是DNase-seq的主要步骤:
-
染色质状态:在最上方,我们看到了细胞核中的染色质。染色质由DNA和与之关联的蛋白质(例如组蛋白)组成。在这里,染色质中的某些区域(由黄色线表示)是开放的,而其他区域是紧密结合的。
-
DNase-I消化:将细胞暴露于DNA酶I (DNase-I) 下。DNase-I是一种可以切割双链DNA的酶,特别在开放的染色质区域活跃。消化的程度取决于DNase-I的浓度和暴露时间。
-
收集DNA:经DNase-I处理后的DNA片段被收集起来。然后,通过大小选择方法,通常选择长度在100-300bp之间的DNA片段。
-
测序:收集的DNA片段被进行高通量测序,如图中所示。这可以产生大量(例如,60-100M)的测序读数。
-
DNase-seq读数计数:最终,测序读数被映射回参考基因组。这生成了一个图谱,表示开放染色质区域的位置和强度。在这里,高的峰代表了高度开放的染色质区域,可能是转录因子结合位点或其他调控元件。
ATAC-seq
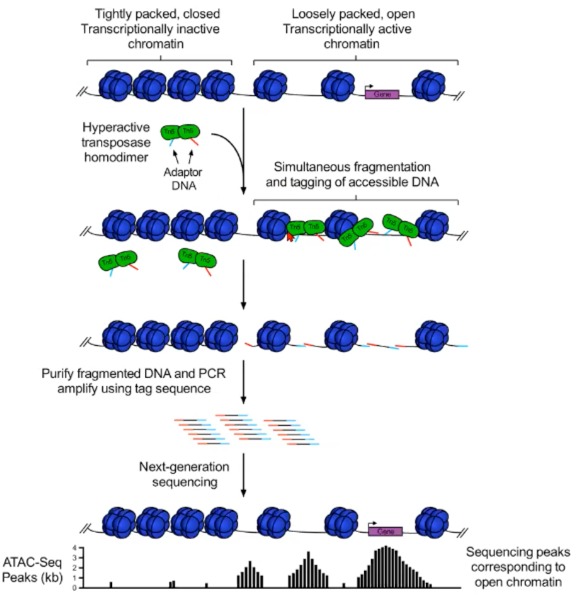
测序测定染色质可及性
ATAC-seq是一种用于鉴定基因组中开放染色质区域的技术,它能够快速、高效地为细胞提供转录调控信息。
以下是ATAC-seq的主要步骤:
-
染色质状态:左边表示紧密打包的、转录非活跃的染色质。右边表示松散打包的、转录活跃的染色质。后者的染色质区域更为“开放”,更易于转录因子和其他蛋白质访问。
-
超活跃转座酶:使用超活跃的转座酶结合适配器DNA对细胞进行处理。转座酶能够同时对DNA进行断裂和标记。
-
同时的DNA断裂和标记:在松散打包的、转录活跃的染色质中,超活跃的转座酶将适配器DNA插入到开放的染色质区域。
-
纯化和PCR扩增:然后从细胞中纯化出已标记的DNA片段,并使用与适配器序列相对应的标签进行PCR扩增。
-
下一代测序:利用下一代测序技术对扩增的DNA片段进行测序。得到的测序读数反映了细胞中开放染色质的位置。
-
ATAC-Seq峰值:最终,使用测序数据在基因组上鉴定开放染色质的区域。这些区域在图上显示为“峰值”,每个峰代表一个开放染色质的位置。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











