







title: T2T基因组组装技术原理
date: 2024-11-21 17:21:37
categories: T2T组装教程
cover: /img/cover2.jpg
tags: T2T
T2T基因组组装技术原理
背景介绍
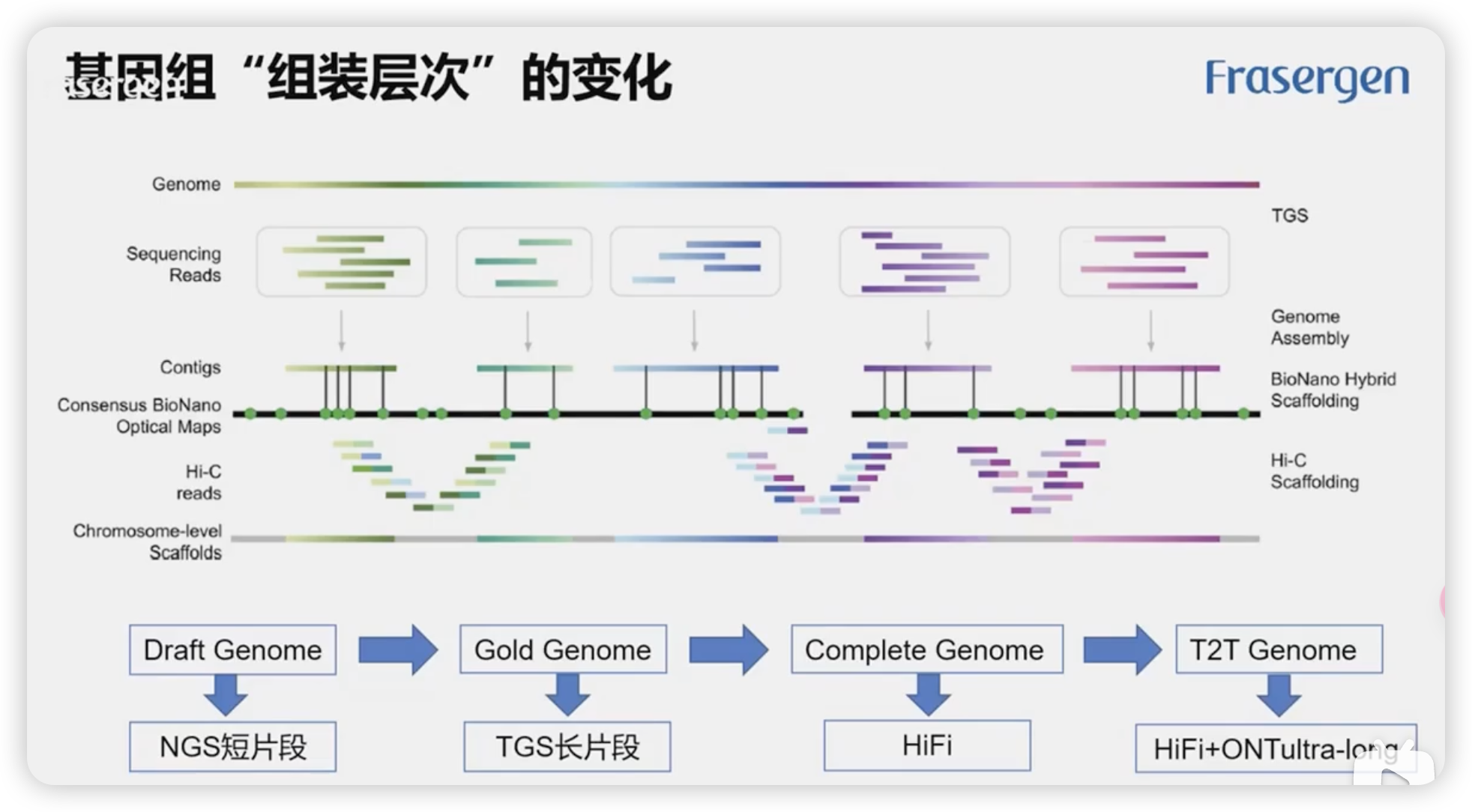
基因组组装
-
Reads(基础原始数据)→ 拼接生成 Contigs(无间隙连续序列)→ 通过连接生成 Scaffolds(更长的框架序列)
-
测序reads
- NGS 下一代测序 短读长 100-300 bp
- TGS 第三代测序 长读长 10-100 kb
-
contigs
- 根据reads重叠拼接成的连续序列片段
-
Scaffolds
- 由多个 contigs 通过额外的信息(如测序技术的配对末端信息paired-end reads、光学图谱、Hi-C 等)链接而成更长的序列片段
- 其中可能包含未知序列(以“N”表示)
-
基因组分类
- Draft Genome(初级基因组):基于 NGS 短读段。
- Gold Genome(金标准基因组):采用 TGS(长读段)。
- Complete Genome(完整基因组):结合 HiFi 技术,质量和精度进一步提高。
- T2T Genome(端到端基因组):结合 HiFi 和超长读段(如 ONT 技术),实现真正无间隙的基因组组装。
-
Hi-C
- Hi-C是一种基于染色质三维结构的高通量染色体构象捕获技术,用于检测DNA在空间中的相互作用。
- 主要用途
- 基因组组装:通过Hi-C可以推断基因组不同片段在染色体中的位置和方向,帮助将contigs组装成染色体级别的scaffolds。
- 三维基因组学:研究基因组的空间结构(如TADs,拓扑关联域)及其与基因调控的关系。
-
HiFi reads
- 提供高精度长读段数据 contigs
-
随着HiFi技术和ONT超长技术的发展
- 可以测得以前的基因组黑洞区域(端粒、着丝粒和高度重复的区域)
T2T基本概念
- 近完成图(Gapless Genome):指在染色体水平组装中,至少有一条染色体由一个单独的 contig 组成,即这条染色体没有任何间隙(gap)。
- 完成图(Gap-free Genome):指染色体上的所有组装区域均为 0 gap(不包括端粒、着丝粒等复杂区域)。
- 例如,若基因组有 12 条染色体,则组装的 contig 也有 12 个,每个 contig 对应一条染色体(如水稻基因组的 12 条染色体)。
- T2T基因组(Telomere-to-Telomere Genome):在完成图的基础上,通过结合 FISH 技术、基于 CenHB3 抗体的 ChIP-seq 着丝粒分离分析等方法,准确鉴定出着丝粒和端粒区域,实现至少一条染色体从端粒到端粒的完整组装。
发表的这些论文中组装出来的T2T染色体占所有的染色体比例
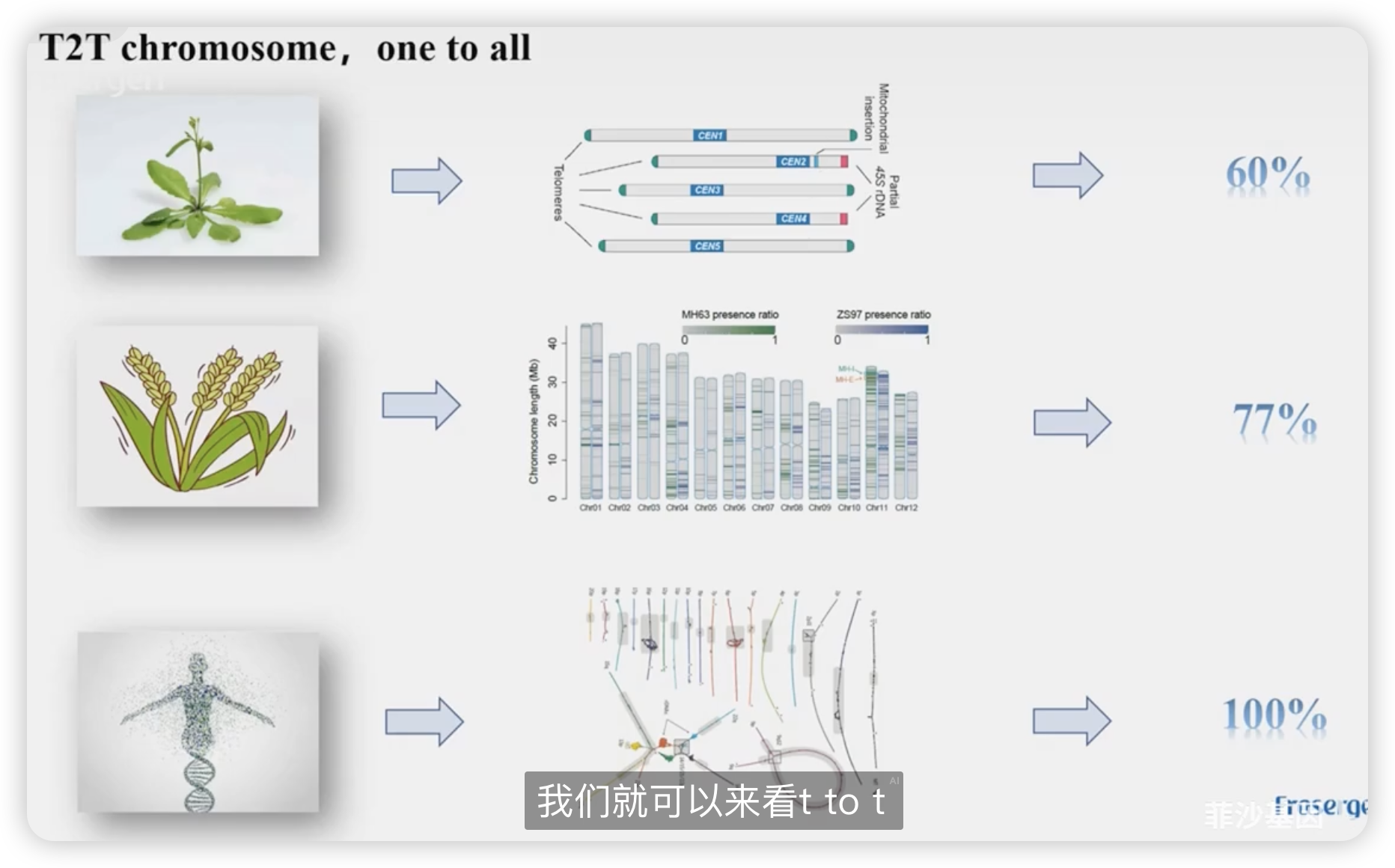
为什要做T2T基因组
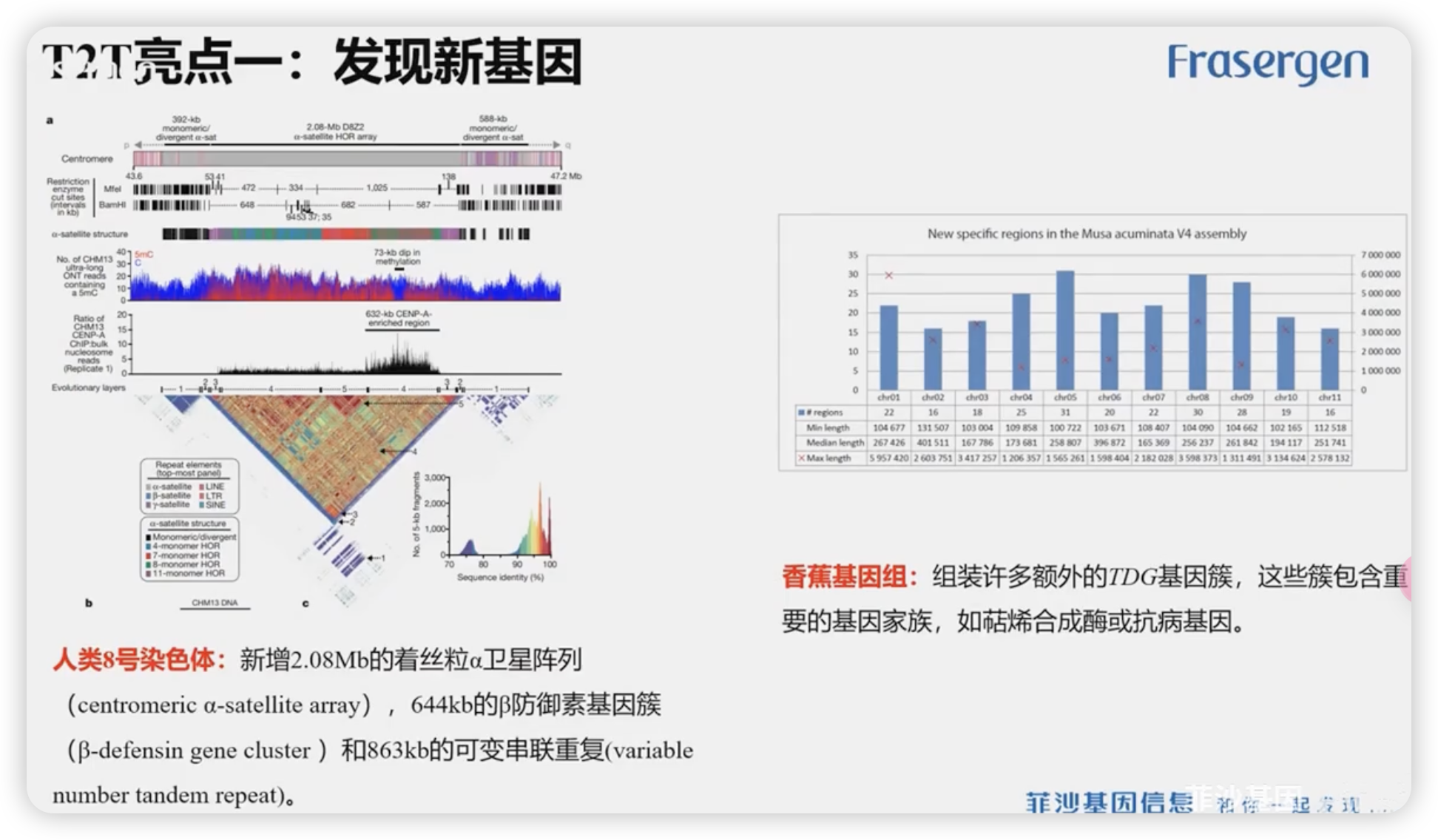
T2T亮点1:发现新基因
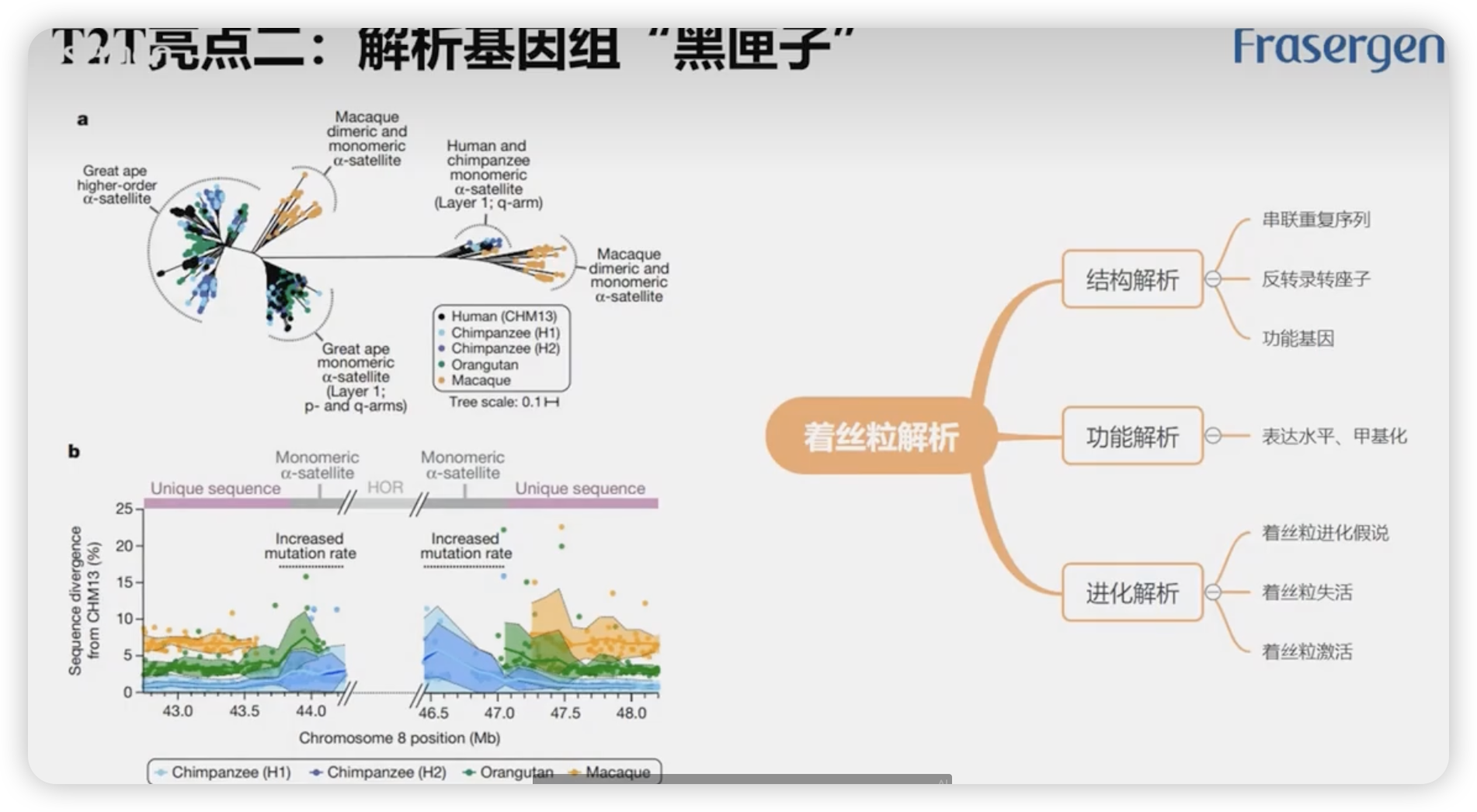
T2T亮点2:解析基因组“黑匣子”
着丝粒这块下功夫去深入研究
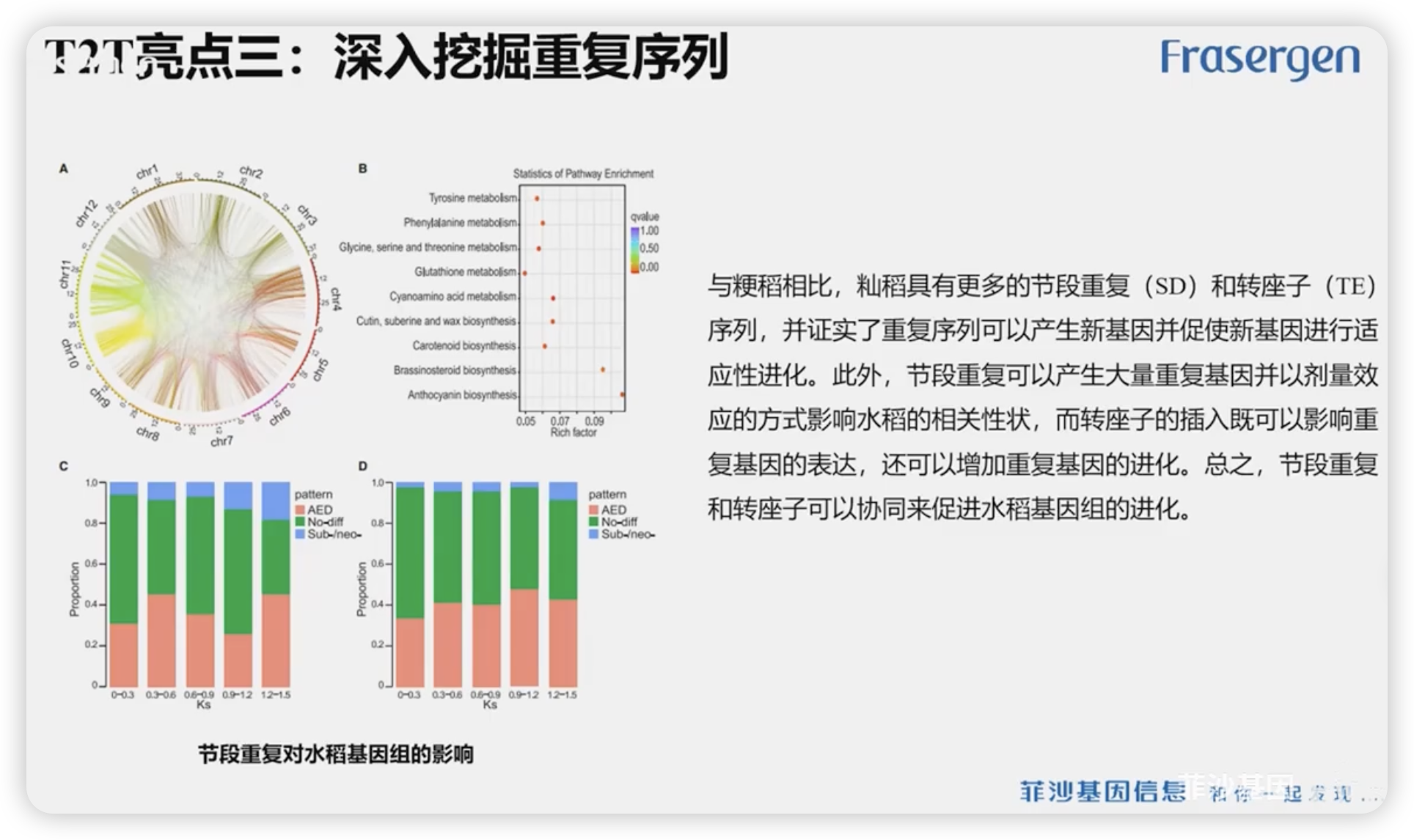
T2T亮点3:深入挖掘重复序列
T2T基因组怎么做
材料的选择
取材非常重要

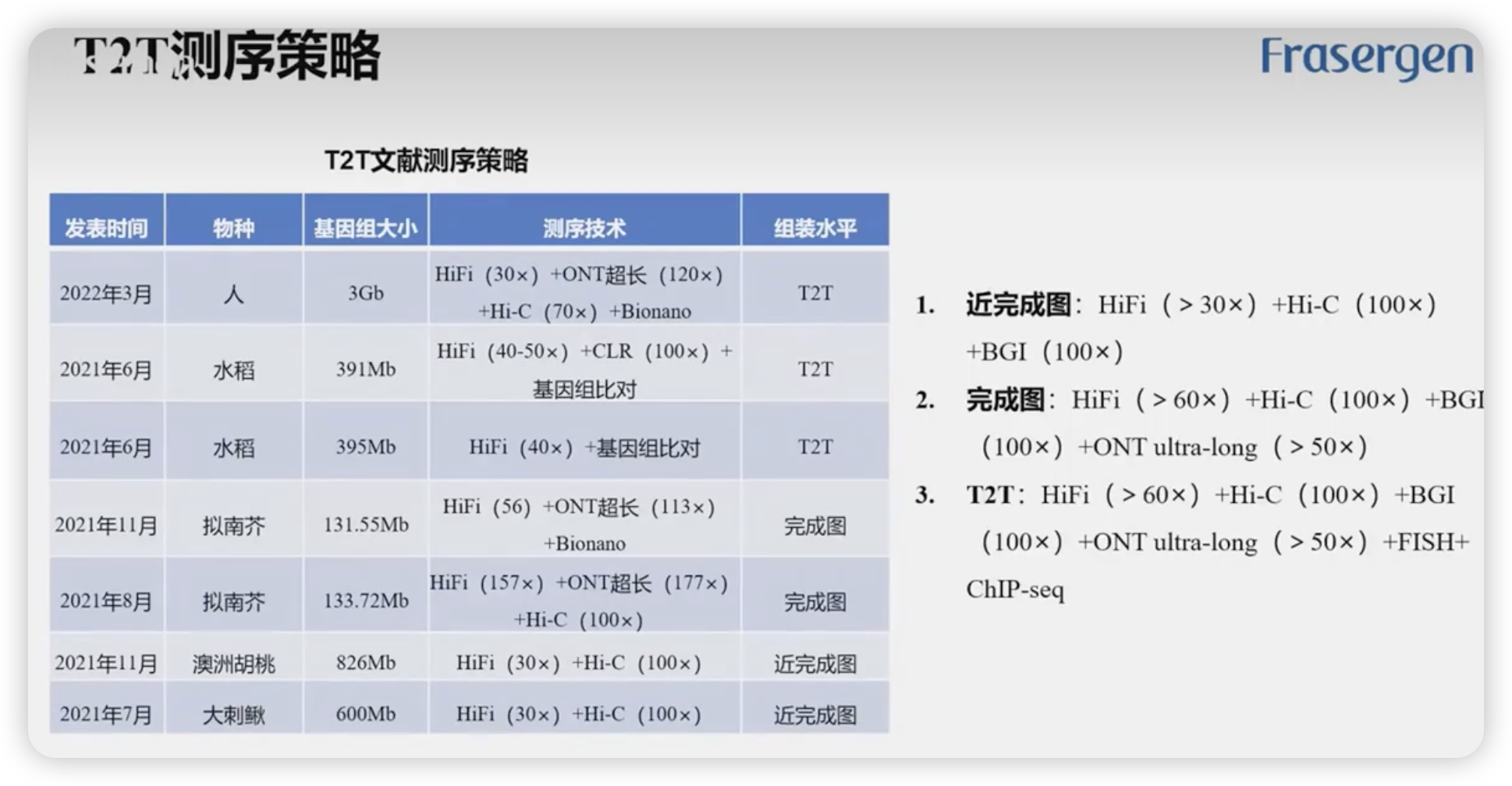
T2T测序策略
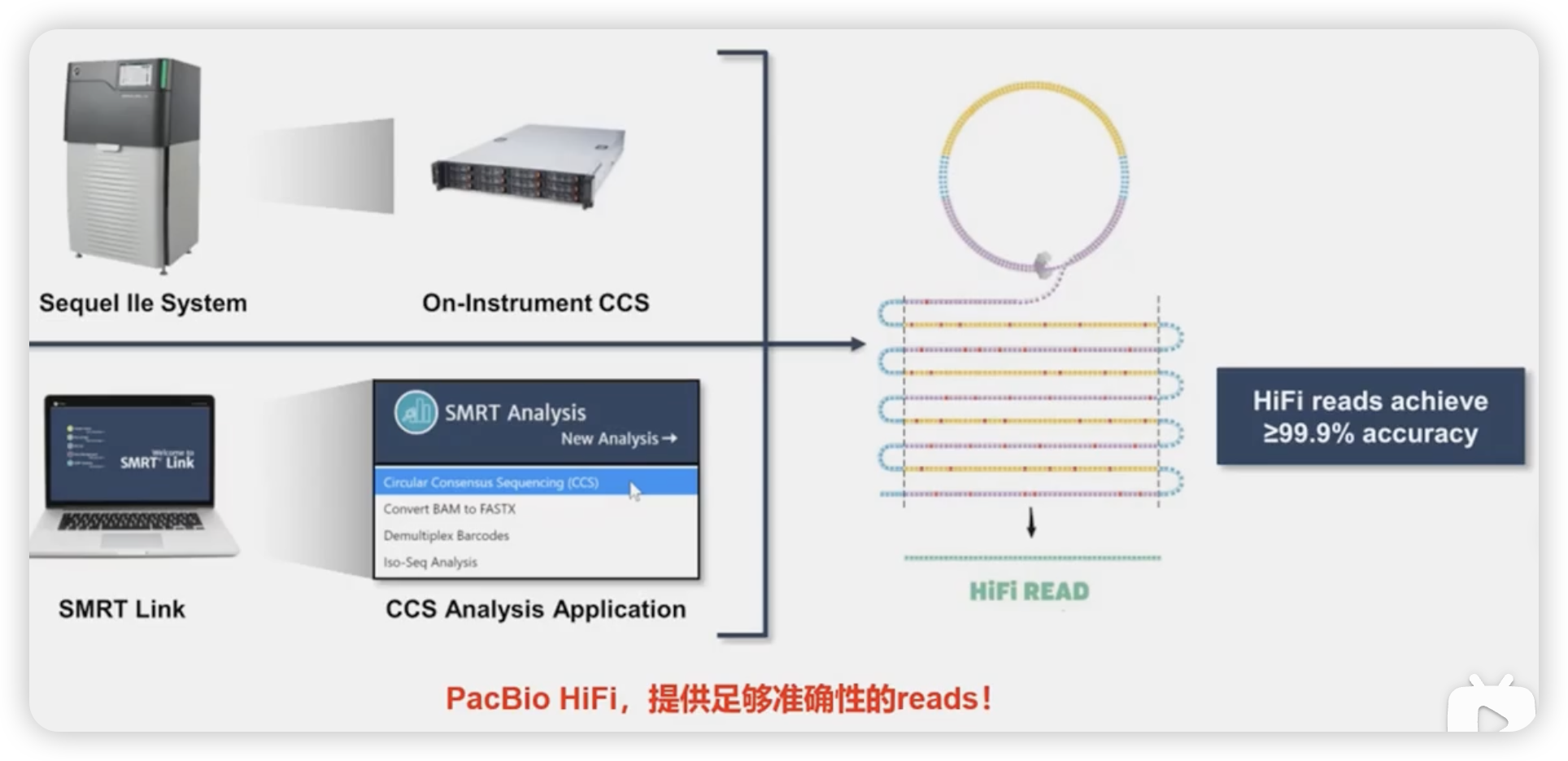
HiFi测序
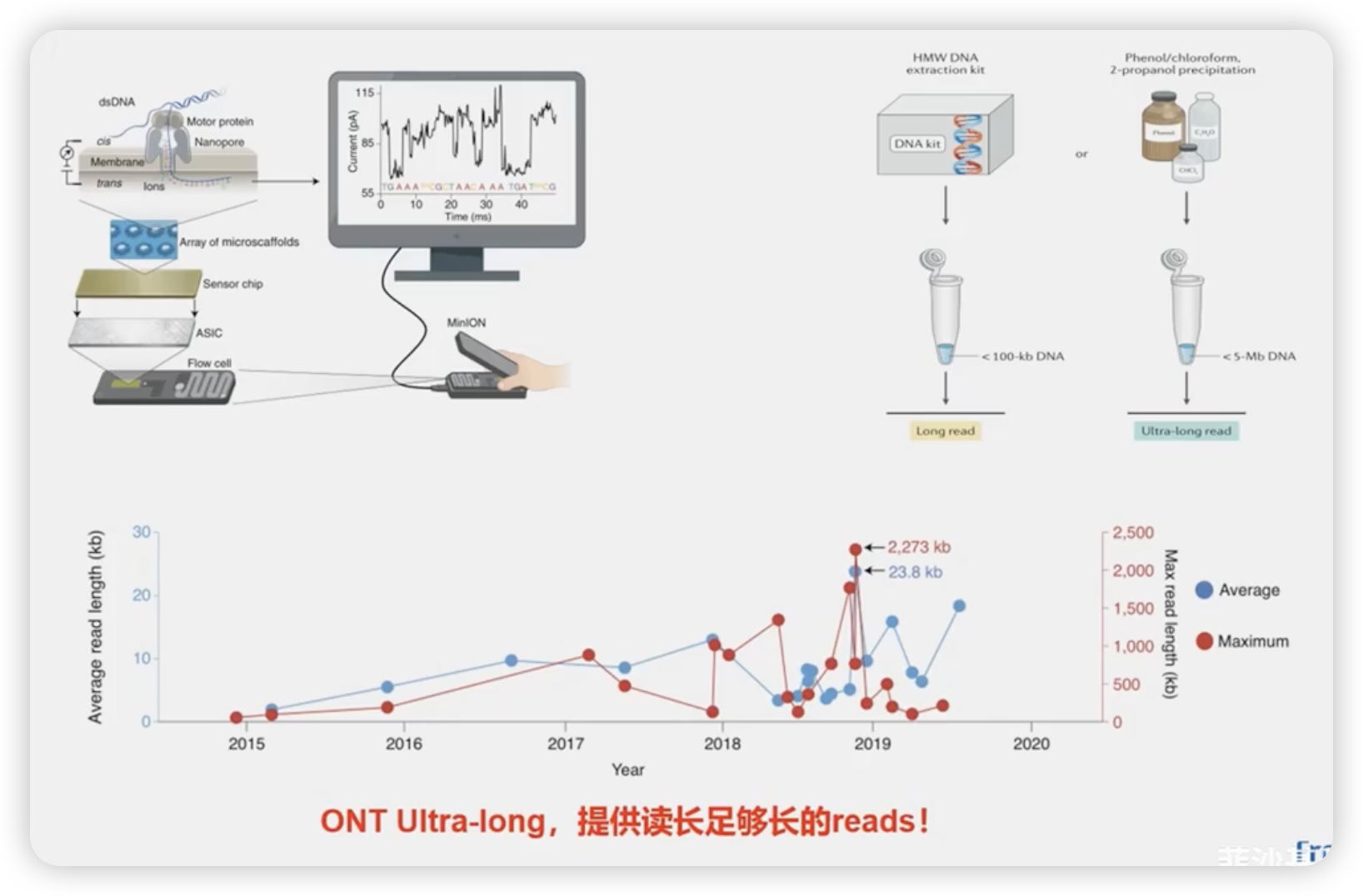
ONT超长测序
- T2T的测序组成
- 测序深度是指基因组上每个碱基被覆盖测序的平均次数。例如:
- 30×:基因组中的每个碱基平均被测序了30次。
- 100×:基因组中的每个碱基平均被测序了100次。
- 测序深度是指基因组上每个碱基被覆盖测序的平均次数。例如:
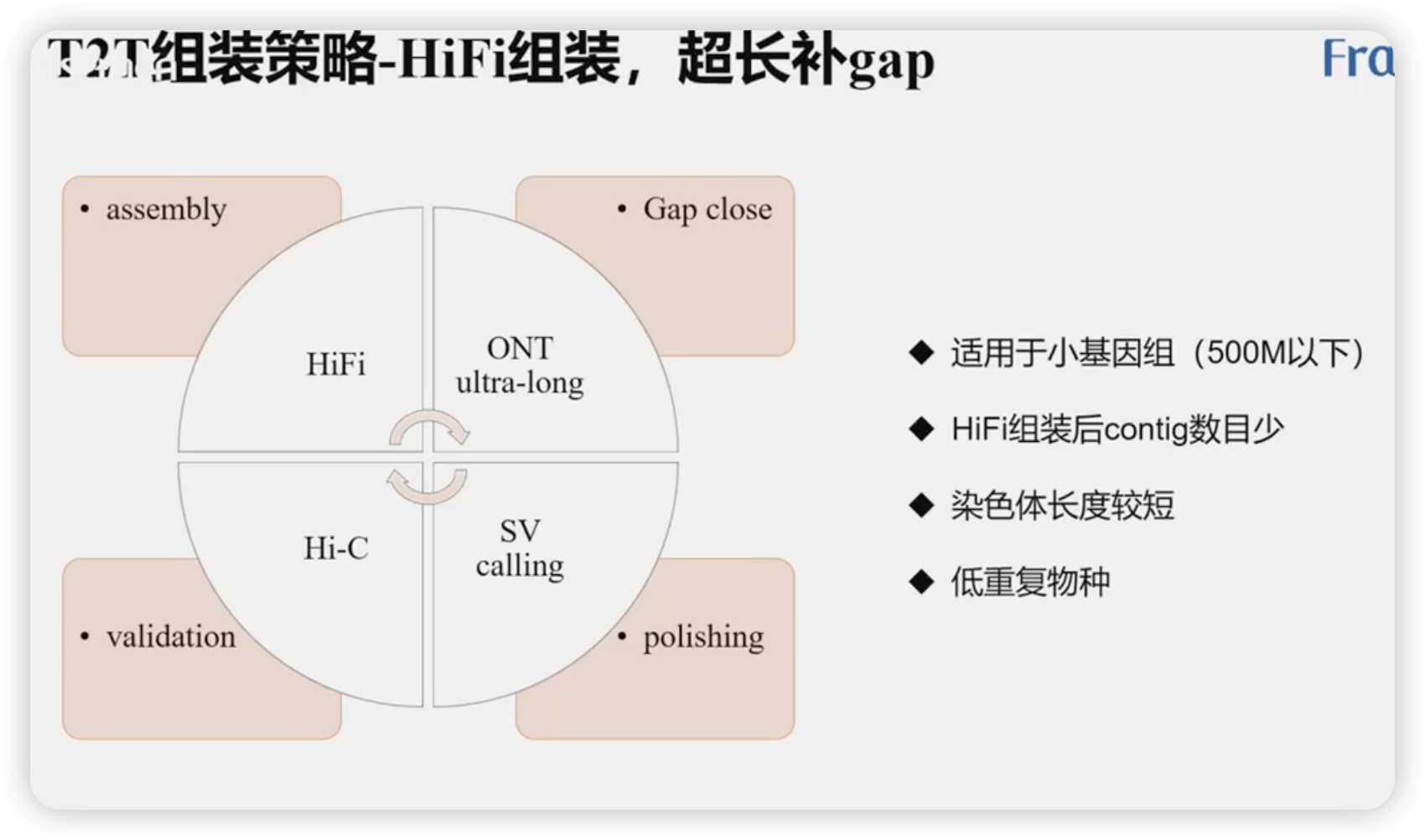
策略1:HiFi组装,ONT超长补gap
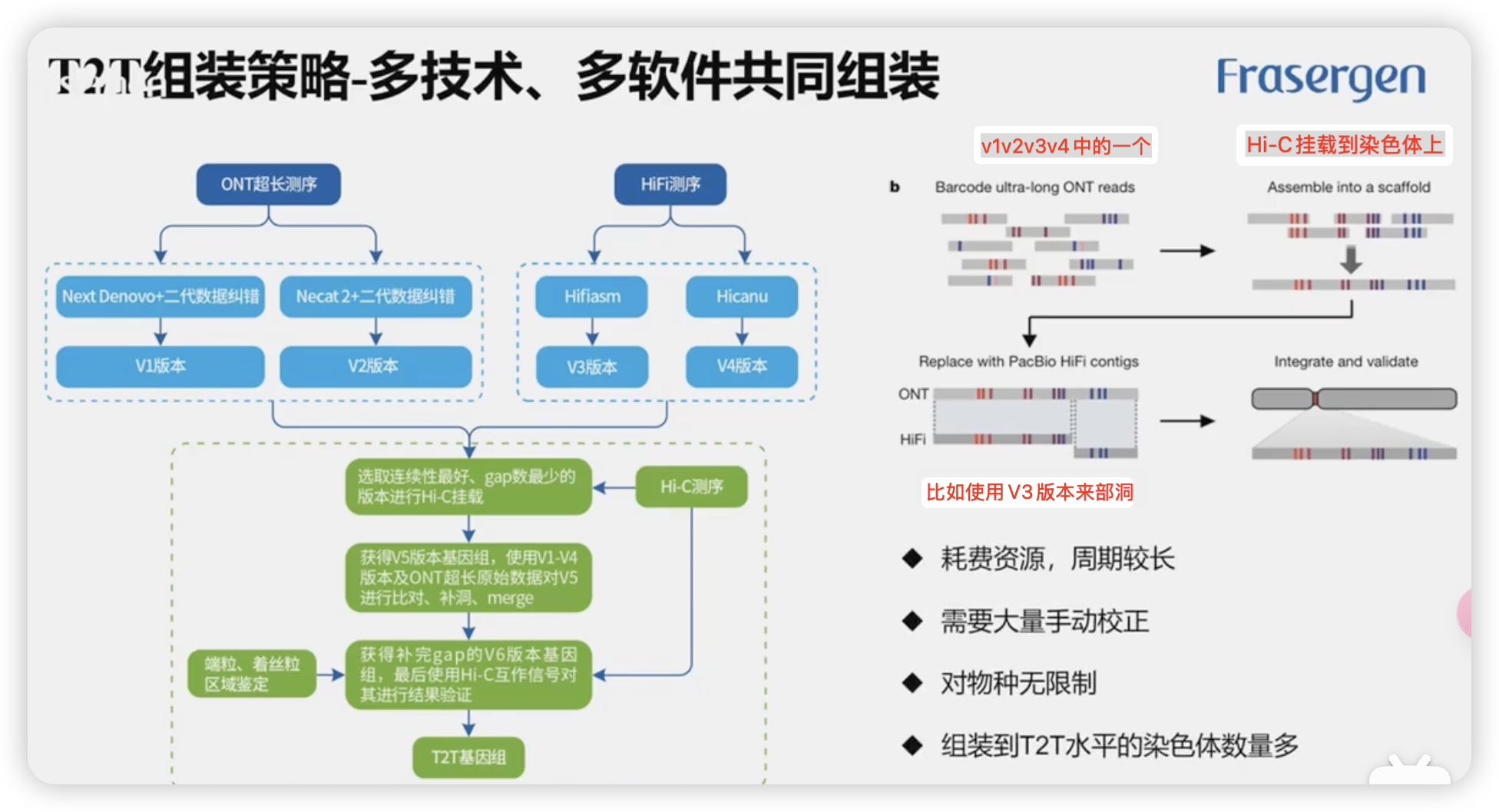
策略2:多技术、多软件共同组装
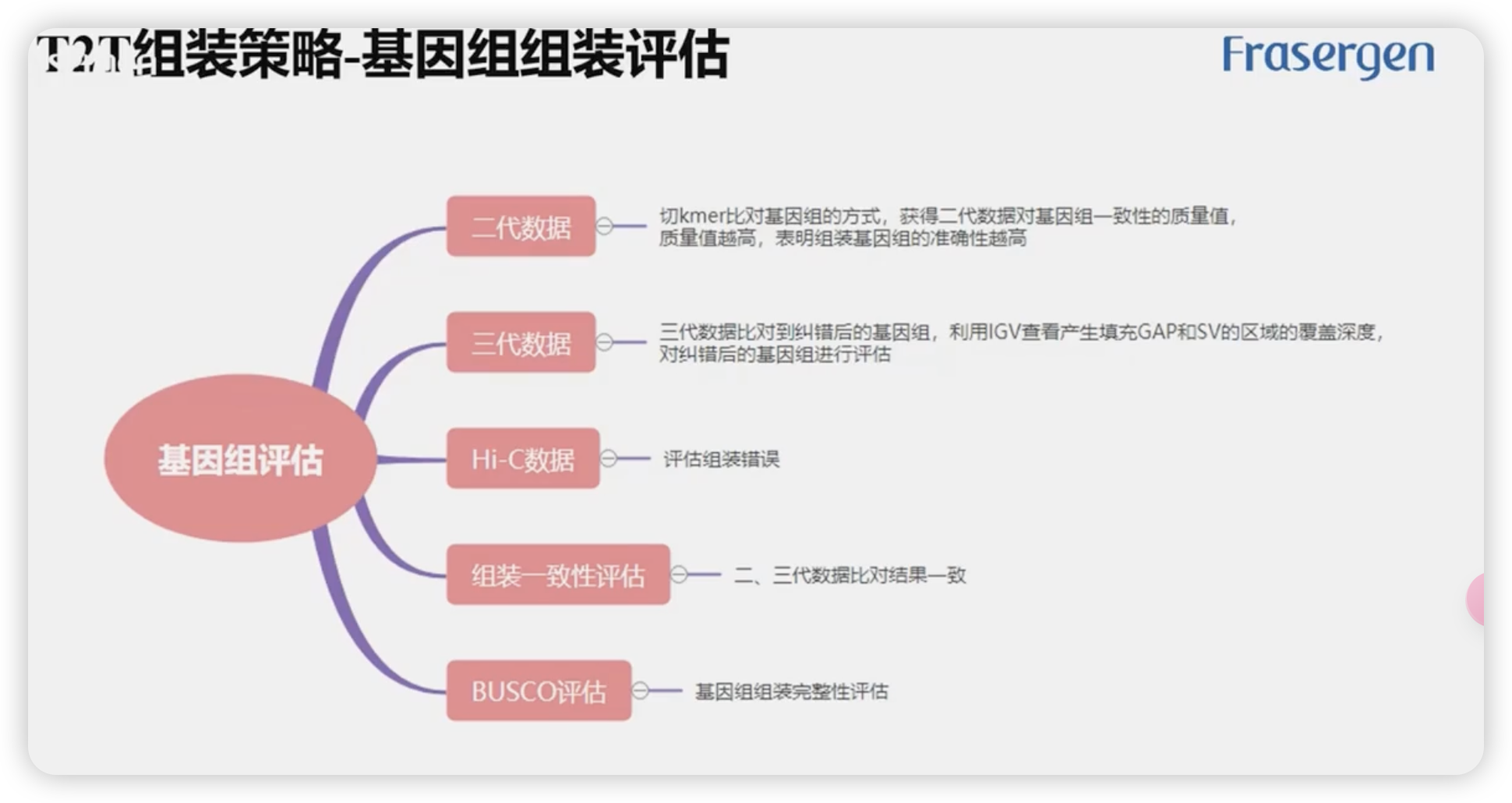
基因组组装评估
基因组组装的评价对T2T基因组组装必不可少
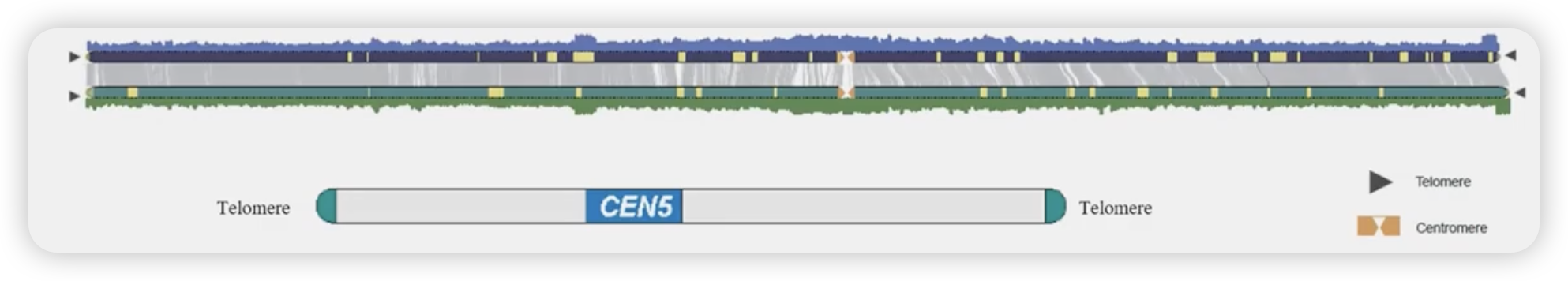
端粒
- 端粒简介
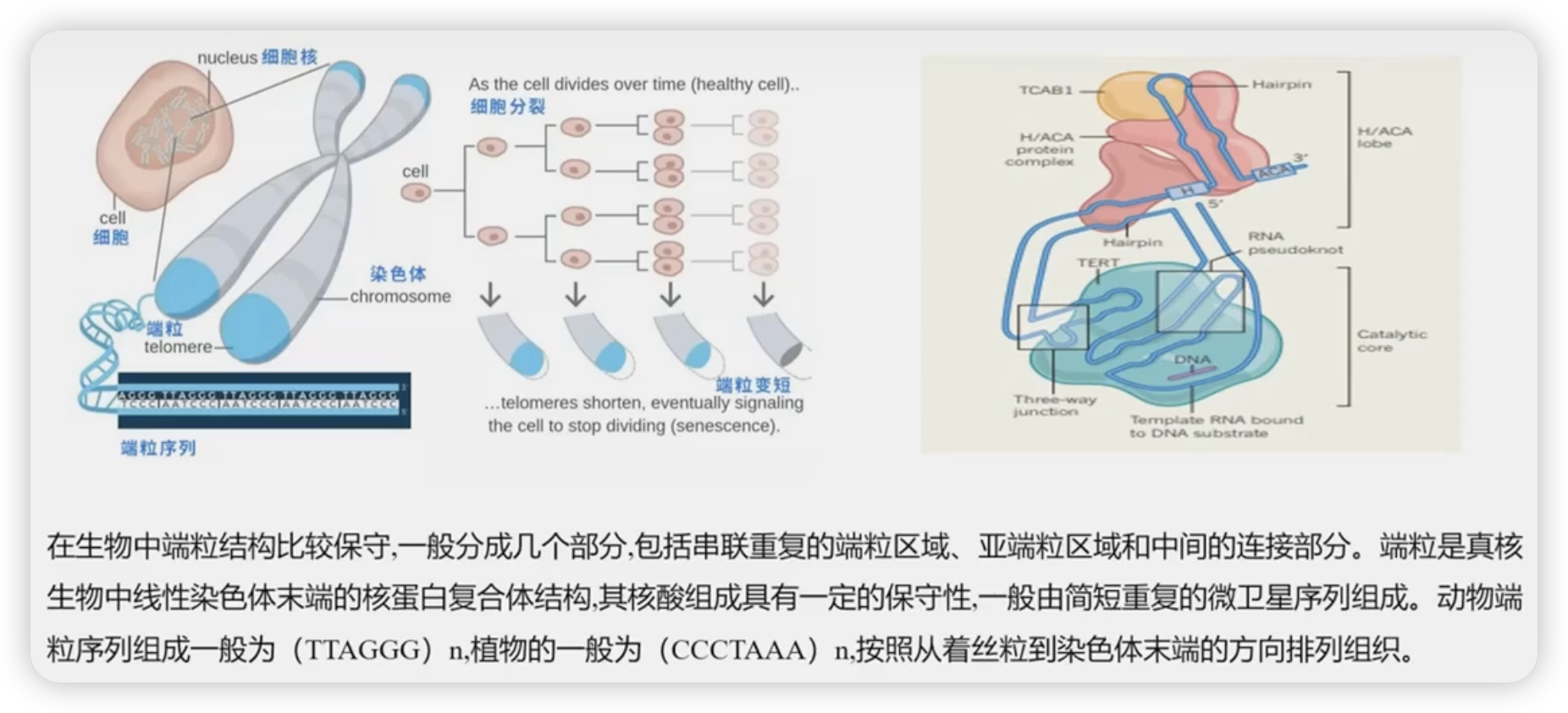
- 端粒鉴定

着丝粒
相对来说,端粒的鉴定较为简单,而着丝粒比较的复杂
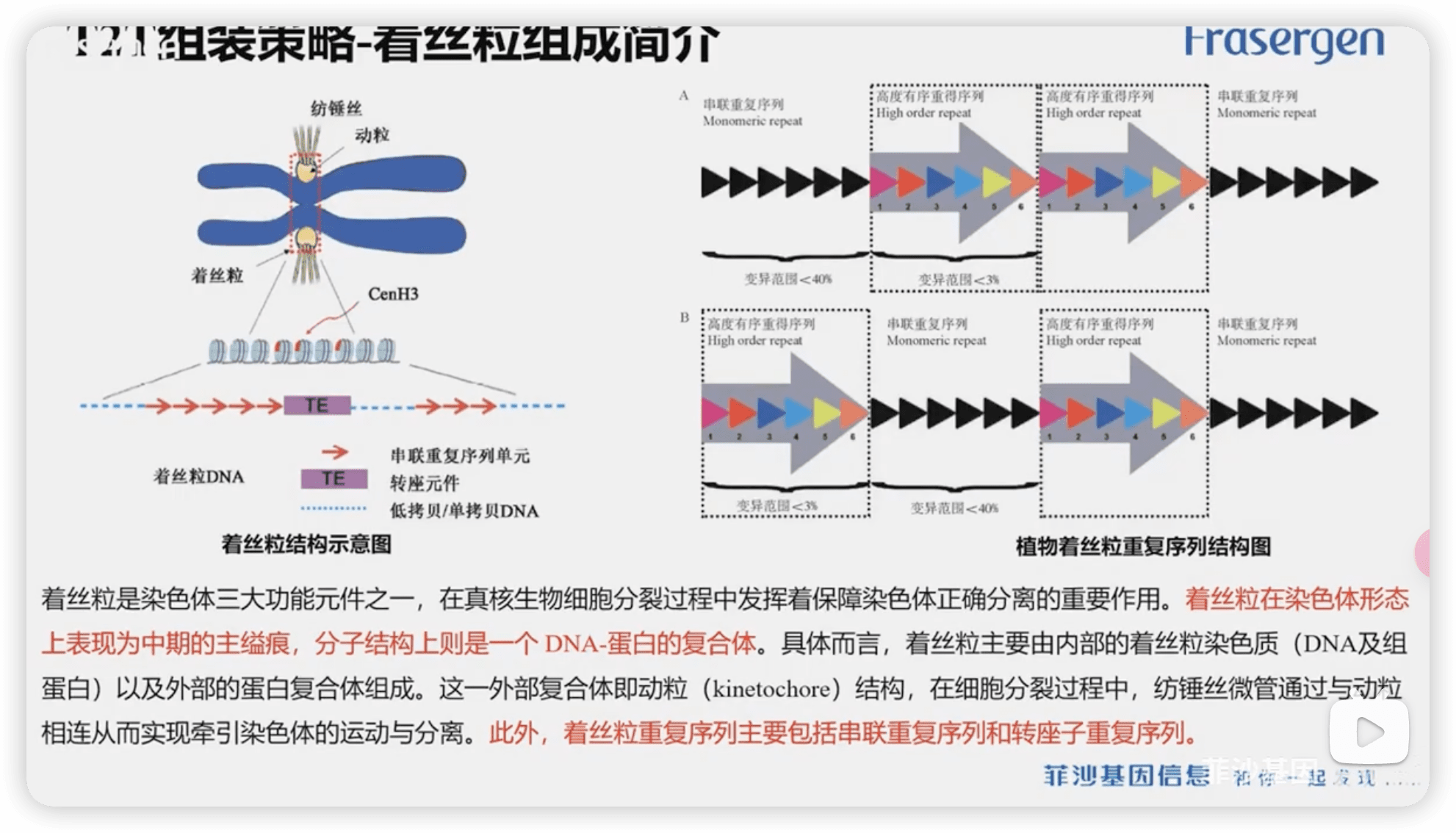
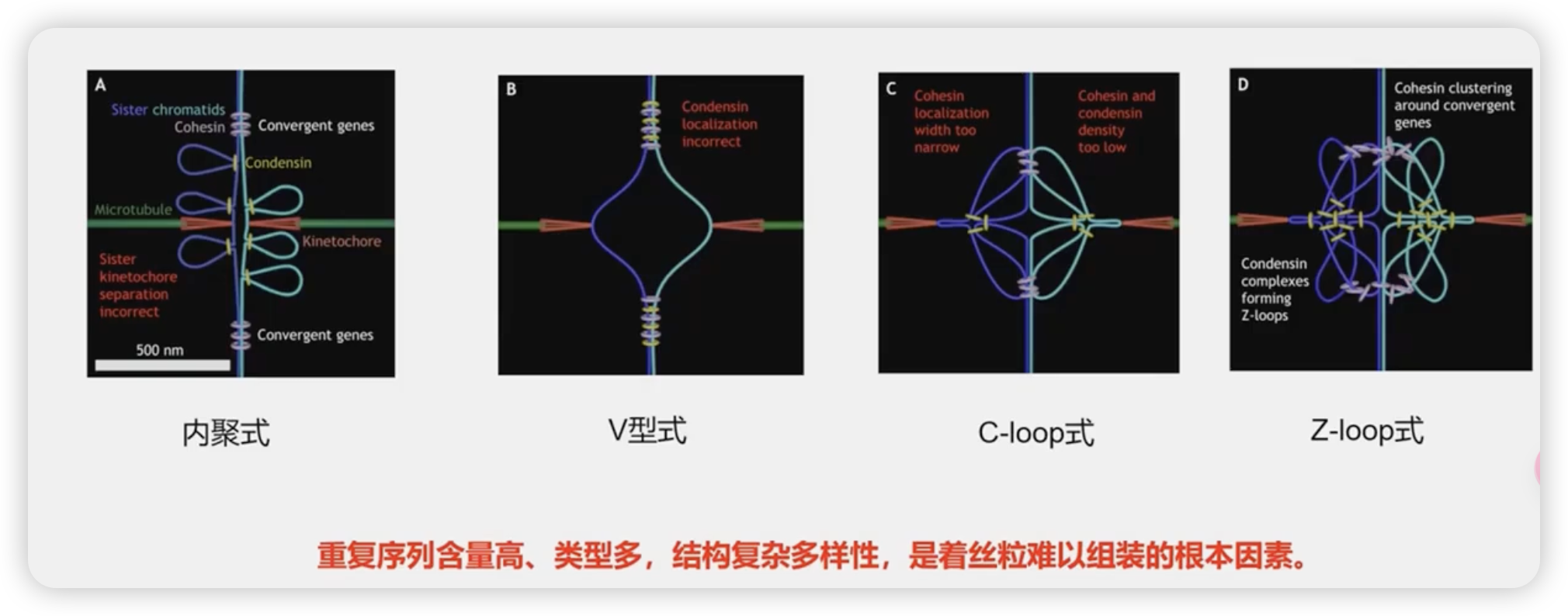

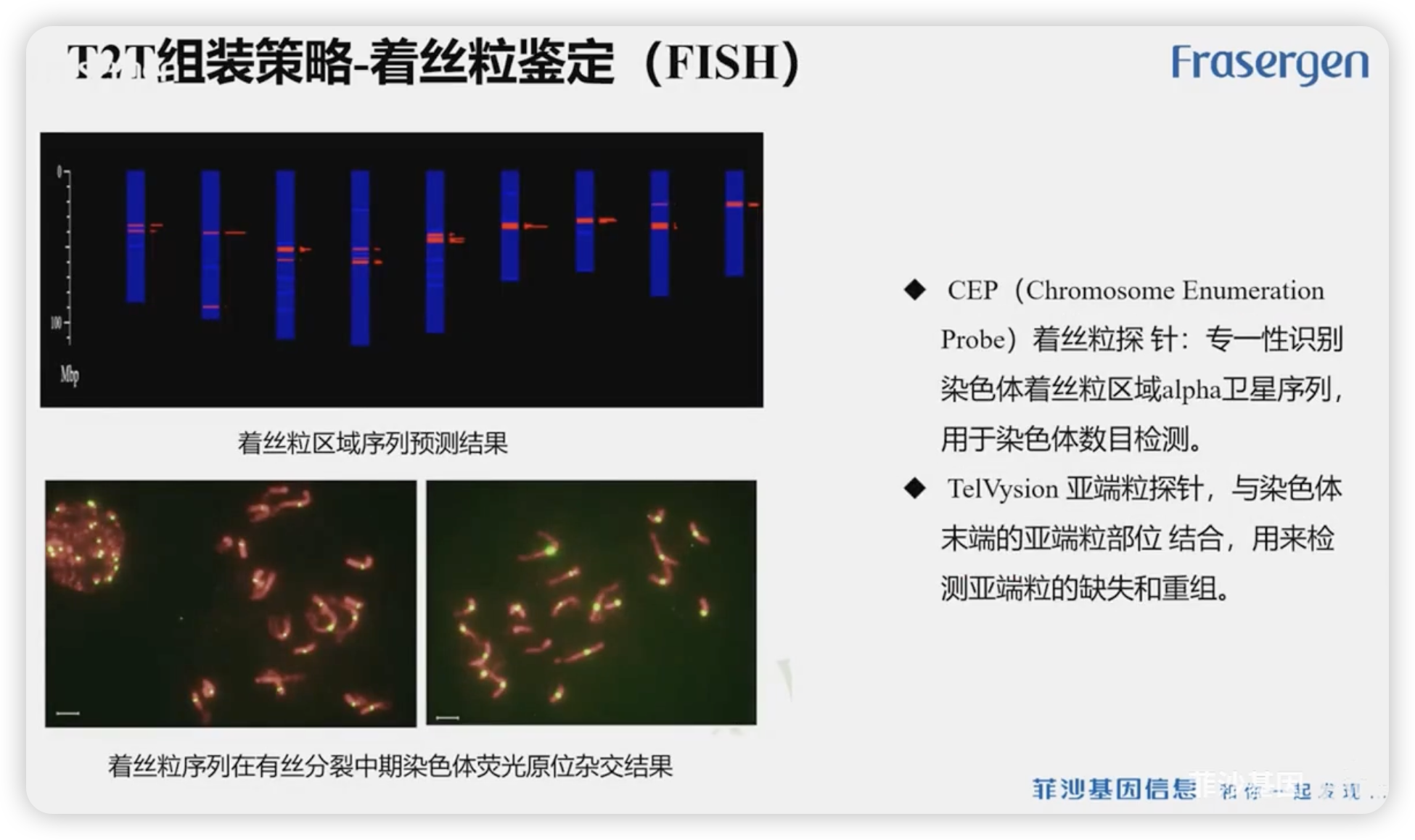


















 1142
1142

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











