







When downloading software, you will often find binaries labeled with either i386 or x86_64. Without going too deep into details, these numbers refer to the type of processor the binaries were compiled for. Luckily, there is an easy way to figure out which type of processor is on the machine you are installing to.
<span style="background-color: rgb(51, 255, 51);">$ uname -a</span>
Linux poset.cgrl.berkeley.edu 2.6.18-238.12.1.el5 #1 SMP Tue May 31 13:22:04 EDT 2011 x86_64 x86_64 x86_64 GNU/Linux
<span style="background-color: rgb(255, 153, 0);">$ unzip bowtie-0.12.7-linux-x86_64.zip</span>
Archive: bowtie-0.12.7-linux-x86_64.zip
creating: bowtie-0.12.7/
creating: bowtie-0.12.7/scripts/
inflating: bowtie-0.12.7/scripts/build_test.sh
inflating: bowtie-0.12.7/scripts/make_a_thaliana_tair.sh
...
...
<span style="background-color: rgb(255, 153, 0);">$ ls -l bowtie-0.12.7/</span>
total 12420
-rw-r--r-- 1 cellison cellison 703 2009-12-12 06:29 AUTHORS
-rwxr-xr-x 1 cellison cellison 773677 2010-09-07 14:20 bowtie
-rwxr-xr-x 1 cellison cellison 327545 2010-09-07 14:19 bowtie-build
-rwxr-xr-x 1 cellison cellison 2683055 2010-09-07 14:19 bowtie-build-debug
-rwxr-xr-x 1 cellison cellison 6930626 2010-09-07 14:19 bowtie-debug
-rwxr-xr-x 1 cellison cellison 238105 2010-09-07 14:19 bowtie-inspect
-rwxr-xr-x 1 cellison cellison 1521358 2010-09-07 14:19 bowtie-inspect-debug
-rw-r--r-- 1 cellison cellison 5207 2008-08-13 09:00 COPYING
drwxr-xr-x 2 cellison cellison 4096 2010-09-07 14:20 doc
drwxr-xr-x 2 cellison cellison 4096 2010-09-07 14:20 genomes
drwxr-xr-x 2 cellison cellison 4096 2011-09-06 10:16 indexes
-rw-r--r-- 1 cellison cellison 69183 2010-09-07 13:54 MANUAL
-rw-r--r-- 1 cellison cellison 80488 2010-09-07 13:47 MANUAL.markdown
-rw-r--r-- 1 cellison cellison 29755 2010-09-07 13:47 NEWS
drwxr-xr-x 2 cellison cellison 4096 2010-09-07 14:20 reads
drwxr-xr-x 3 cellison cellison 4096 2010-09-07 14:20 scripts
-rw-r--r-- 1 cellison cellison 6258 2009-10-05 14:55 TUTORIAL
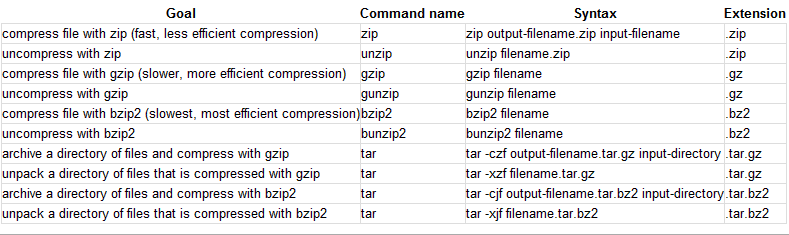
-rw-r--r-- 1 cellison cellison 6 2010-09-07 13:47 VERSIONCheatsheet showing various methods for compressing/uncompressing files and packaging/unpackaging directories
zcat命令用于不真正解压缩文件,就能显示压缩包中文件的内容的场合
-c将文件内容写到标准输出
echo命令用来打印信息,是一个最常用的命令。在命令行中常用来打印环境变量的值,已确定当前环境中是否设置了指定的环境变量。在shell脚本中,常用来打印信息和帮助调试程序。
zcat my.fastq.gz | echo $((`wc -l`/4))
(..) means the parent of the current directory, so typing
% cd ..will take you one directory up the hierarchy
Note: typing cd with no argument always returns you to your home directory. This is very useful if you are lost in the file system.
Home directories can also be referred to by the tilde ~ character. It can be used to specify paths starting at your home directory. So typing
% ls ~/unixstuff
Copy data directory into your home directory
cp r ~panu/tuesday ~-r :遞迴持續複製,用於目錄的複製行為;(常用)
Extract fields 2, 4, and 5 from file.txt:
awk '{print $2,$4,$5}' input.txtPrint each line where the 5th field is equal to ‘abc123’:
awk '$5 == "abc123"' file.txtPrint each line whose 7th field does not match the regular expression:
awk '$7 !~ /^[a-f]/' file.txtGet unique entries in file.txt based on column 2 (takes only the first instance):
awk '!arr[$2]++' file.txtPrint rows where column 3 is larger than column 5 in file.txt:
awk '$3>$5' file.txtSum column 1 of file.txt:
awk '{sum+=$1} END {print sum}' file.txtCompute the mean of column 2:
awk '{x+=$2}END{print x/NR}' file.txtPrint everything except the first line:
awk 'NR>1' input.txtPrint rows 20-80:
awk 'NR>=20&&NR<=80' input.txtSearch for .bam files anywhere in the current directory recursively:
find . -name "*.bam"Delete all .bam files (Irreversible: use with caution! Confirm list BEFORE deleting):
find . -name "*.bam" | xargs rmDetermine the number of genes annotated in a GFF3 file.
grep -c $'\tgene\t' yourannots.gff3Extract all gene IDs from a GFF3 file.
grep $'\tgene\t' yourannots.gff3 | perl -ne '/ID=([^;]+)/ and printf("%s\n", $1)'Print length of each gene in a GFF3 file.
grep $'\tgene\t' yourannots.gff3 | cut -s -f 4,5 | perl -ne '@v = split(/\t/); printf("%d\n", $v[1] - $v[0] + 1)'get the sequences length distribution form a fastq file using awk
zcat file.fastq.gz | awk 'NR%4 == 2 {lengths[length($0)]++} END {for (l in lengths) {print l, lengths[l]}}'Reverse complement a sequence (I use that a lot when I need to design primers)
echo 'ATTGCTATGCTNNNT' | rev | tr 'ACTG' 'TGAC'awk 'BEGIN{RS=">";FS="\n"}NR>1{if ($1~/Medtr1g006/) print ">"$0}' fasta.txt###Awk one-liners for FASTA manipulation
###Created by Melissa M.L. Wong (melissawongukm@gmail.com) on 6th July 2015
###1. To find sequences with matching name
awk 'BEGIN{RS=">";FS="\n"}NR>1{if ($1~/Medtr1g006/) print ">"$0}' fasta.txt
###2. To extract sequences using a list
awk 'BEGIN{RS=">";FS="\n"}NR>1{if ($1~/Medtr1g006/) print ">"$1}' fasta.txt
#This creates the file "list.txt". It contains names which start with ">" and each name is separated by a newline ("\n").
awk 'BEGIN{RS=">";FS="\n"}NR==FNR{a[$1]++}NR>FNR{if ($1 in a && $0!="") printf ">%s",$0}' list.txt fasta.txt
###3. To join multiple lines into single line
awk 'BEGIN{RS=">";FS="\n"}NR>1{seq="";for (i=2;i<=NF;i++) seq=seq""$i; print ">"$1"\n"seq}' fasta.txt
#Single line sequence is desirable when a sequence is long and spans many lines. Furthermore, single line sequence is much easier to be manipulated using AWK oneliners as showed in the next few examples.
###4. To print specified sequence region
#To print the sequence starting from position 1 until 221
awk 'BEGIN{RS=">";FS="\n"}NR>1{seq="";for (i=2;i<=NF;i++) seq=seq""$i; print ">"$1"\n"substr(seq,1,221)}' fasta.txt
#To print sequence starting from position 10 until 90
awk 'BEGIN{RS=">";FS="\n"}NR>1{seq="";for (i=2;i<=NF;i++) seq=seq""$i; print ">"$1"\n"substr(seq,10,90-10+1)}' fasta.txt
#To print sequence with matching name from position 10 until 90
awk 'BEGIN{RS=">";FS="\n"}NR>1{seq="";for (i=2;i<=NF;i++) seq=seq""$i; if ($1~/Medtr1g006/) print ">"$1"\n"substr(seq,10,90-10+1)}' fasta.txt
#Useful to print sequence region when given start position and stop position or length
###5. To reformat sequences into 100 characters per line
awk 'BEGIN{RS=">";FS="\n"}NR>1{seq="";for (i=2;i<=NF;i++) seq=seq""$i;a[$1]=seq;b[$1]=length(seq)}END{for (i in a) {k=sprintf("%d", (b[i]/100)+1); printf ">%s\n",i;for (j=1;j<=int(k);j++) printf "%s\n", substr(a[i],1+(j-1)*100,100)}}' fasta.txt
###6. To substitute nucleotide sequences
#To substitute capital letter with small letter
awk 'BEGIN{RS=">";FS="\n"}NR>1{printf ">%s",$1;for (i=2;i<=NF;i++) printf "\n%s",tolower($i)}' fasta.txt
###7. To convert DNA to RNA
awk 'BEGIN{RS=">";FS="\n"}NR>1{printf ">%s\n",$1;for (i=2;i<=NF;i++) {gsub(/T/,"U",$i); printf "%s\n",$i}}' fasta.txt
###8. To summarize sequence content
awk 'BEGIN{RS=">";FS="\n";print "name\tA\tC\tG\tT\tN\tlength\tGC%"}NR>1{sumA=0;sumT=0;sumC=0;sumG=0;sumN=0;seq="";for (i=2;i<=NF;i++) seq=seq""$i;k=length(seq); for (i=1;i<=k;i++) {if (substr(seq,i,1)=="T") sumT+=1; else if (substr(seq,i,1)=="A") sumA+=1; else if (substr(seq,i,1)=="G")sumG+=1; else if (substr(seq,i,1)=="C") sumC+=1; else if (substr(seq,i,1)=="N") sumN+=1}; print $1"\t"sumA"\t"sumC"\t"sumG"\t"sumT"\t"sumN"\t"k"\t"(sumC+sumG)/k*100}' fasta.txt
#Calculate number of each nucleotide, total length and GC content
###9. To reverse complement nucleotide sequences
awk 'BEGIN{RS=">";FS="\n";a["T"]="A";a["A"]="T";a["C"]="G";a["G"]="C";a["N"]="N"}NR>1{for (i=2;i<=NF;i++) seq=seq""$i;for(i=length(seq);i!=0;i--){k=substr(seq,i,1);x=x a[k]}; printf ">%s\n%s\n",$1,x}' fasta.txt
#This will produce a single line sequence
###10. To convert FASTQ to FASTA format
awk 'NR%4==1{print ">"substr($0,2)}NR%4==2{print $0}' fastq.txt
#print first and second line of every four lines. Replace the first character of the first line with ">".














 2466
2466

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









