






eukcc folder
–out outfolder
–threads 8
–links linktable.csv
binfolder
EukCC 首先将分别对所有bins进行运行。随后,它会识别那些至少达到50%完整度但尚未超过100-improve\_percent的中等质量bins。接下来,它会找出那些通过至少100对端读配对与这些中等质量bins相连接的bins。若经过合并后bin的质量评分有所提高,则该bin将会被合并。
已合并的bins可以在输出文件夹中找到。
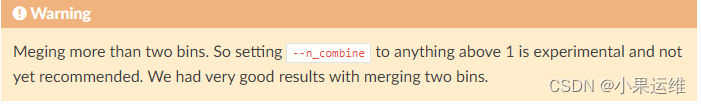
### 警示
blinks.py
#!/usr/bin/env python3
import pysam
from Bio import SeqIO
from collections import defaultdict
import os
import argparse
import logging
import csv
def is_in(read, contig_map, within=1000):
if read.reference_name not in contig_map.keys():
return False
if read.reference_start <= within or read.reference_end <= within:
return True
elif read.reference_start > (
contig_map[read.reference_name] - within
) or read.reference_end > (contig_map[read.reference_name] - within):
return True
else:
return False
def keep_read(read, contig_map, within=1000, min_ANI=98, min_cov=0):
ani = (
(read.query_alignment_length - read.get_tag(“NM”))
/ float(read.query_alignment_length)
* 100
)
cov = read.query_alignment_length / float(read.query_length) * 100
if ani >= min_ANI and cov >= min_cov and is_in(read, contig_map, within) is True:
return True
else:
return False
def contig_map(bindir, suffix=“.fa”):
m = {}
for f in os.listdir(bindir):
if f.endswith(suffix) is False:
continue
path = os.path.join(bindir, f)
with open(path, “r”) as handle:
for record in SeqIO.parse(handle, “fasta”):
m[record.name] = len(record.seq)
return m
def bin_map(bindir, suffix=“.fa”):
contigs = defaultdict(str)
contigs_per_bin = defaultdict(int)
for f in os.listdir(bindir):
if f.endswith(suffix) is False:
continue
path = os.path.join(bindir, f)
binname = os.path.basename(f)
with open(path, “r”) as handle:
for record in SeqIO.parse(handle, “fasta”):
contigs[record.name] = binname
contigs_per_bin[binname] += 1
return contigs, contigs_per_bin
def read_pair_generator(bam):
“”"
Generate read pairs in a BAM file or within a region string.
Reads are added to read_dict until a pair is found.
From: https://www.biostars.org/p/306041/
“”"
read_dict = defaultdict(lambda: [None, None])
for read in bam.fetch():
if not read.is_paired or read.is_secondary or read.is_supplementary:
continue
qname = read.query_name
if qname not in read_dict:
if read.is_read1:
read_dict[qname][0] = read
else:
read_dict[qname][1] = read
else:
if read.is_read1:
yield read, read_dict[qname][1]
else:
yield read_dict[qname][0], read
del read_dict[qname]
def read_bam_file(bamf, link_table, cm, within, ANI):
samfile = pysam.AlignmentFile(bamf, “rb”)
# generate link table
logging.info("Parsing Bam file. This can take a few moments")
for read, mate in read_pair_generator(samfile):
if keep_read(read, cm, within, min_ANI=ANI) and keep_read(
mate, cm, within, min_ANI=ANI
):
# fill in the table
link_table[read.reference_name][mate.reference_name] += 1
if read.reference_name != mate.reference_name:
link_table[mate.reference_name][read.reference_name] += 1
return link_table
def main():
# set arguments
# arguments are passed to classes
parser = argparse.ArgumentParser(
description=“Evaluate completeness and contamination of a MAG.”
)
parser.add_argument(“bindir”, type=str, help=“Run script on these bins”)
parser.add_argument(
“bam”,
type=str,
help=“Bam file(s) with reads aligned against all contigs making up the bins”,
nargs=“+”,
)
parser.add_argument(
“–out”,
“-o”,
type=str,
required=False,
help=“Path to output table (Default: links.csv)”,
default=“links.csv”,
)
parser.add_argument(
“–ANI”, type=float, required=False, help=“ANI of matching read”, default=99
)
parser.add_argument(
“–within”,
type=int,
required=False,
help=“Within this many bp we need the read to map”,
default=1000,
)
parser.add_argument(
“–contigs”,
“-c”,
action=“store_true”,
default=False,
help=“Instead of bins print contigs”,
)
parser.add_argument(
“–quiet”,
“-q”,
dest=“quiet”,
action=“store_true”,
default=False,
help=“Silcence most output”,
)
parser.add_argument(
“–debug”,
“-d”,
action=“store_true”,
default=False,
help=“Debug and thus ignore safety”,
)
args = parser.parse_args()
# define logging
logLevel = logging.INFO
if args.quiet:
logLevel = logging.WARNING
elif args.debug:
logLevel = logging.DEBUG
logging.basicConfig(
format="%(asctime)s %(message)s",
datefmt="%d-%m-%Y %H:%M:%S: ",
level=logLevel,
)
bindir = args.bindir
cm = contig_map(bindir)
bm, contigs_per_bin = bin_map(bindir)
logging.debug("Found {} contigs".format(len(cm)))
link_table = defaultdict(lambda: defaultdict(int))
bin_table = defaultdict(lambda: defaultdict(int))
# iterate all bam files
for bamf in args.bam:
link_table = read_bam_file(bamf, link_table, cm, args.within, args.ANI)
logging.debug("Created link table with {} entries".format(len(link_table)))
# generate bin table
for contig_1, dic in link_table.items():
for contig_2, links in dic.items():
bin_table[bm[contig_1]][bm[contig_2]] += links
logging.debug("Created bin table with {} entries".format(len(bin_table)))
out_data = []
logging.debug("Constructing output dict")
if args.contigs:
for contig_1, linked in link_table.items():
for contig_2, links in linked.items():
out_data.append(
{
"bin_1": bm[contig_1],
"bin_2": bm[contig_2],
"contig_1": contig_1,
"contig_2": contig_2,
"links": links,
"bin_1_contigs": contigs_per_bin[bm[contig_1]],
"bin_2_contigs": contigs_per_bin[bm[contig_2]],
}
)
else:
for bin_1, dic in bin_table.items():
for bin_2, links in dic.items():
out_data.append({"bin_1": bin_1, "bin_2": bin_2, "links": links})
logging.debug("Out data has {} rows".format(len(out_data)))
# results
logging.info("Writing output")
with open(args.out, "w") as fout:
if len(out_data) > 0:
cout = csv.DictWriter(fout, fieldnames=list(out_data[0].keys()))
cout.writeheader()
for row in out_data:
cout.writerow(row)
else:
logging.warning("No rows to write")
if name == “main”:
main()
scripts/filter\_euk\_bins.py
#!/usr/bin/env python3
This file is part of the EukCC (https://github.com/openpaul/eukcc).
Copyright © 2019 Paul Saary
This program is free software: you can redistribute it and/or modify
it under the terms of the GNU General Public License as published by
the Free Software Foundation, version 3.
This program is distributed in the hope that it will be useful, but
WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU
General Public License for more details.
You should have received a copy of the GNU General Public License
along with this program. If not, see http://www.gnu.org/licenses/.
provides all file operation functions
used inthis package
import os
import argparse
import subprocess
import logging
import tempfile
import gzip
from multiprocessing import Pool
backup fasta handler, so we can use readonly directories
class fa_class:
def init(self, seq, name, long_name):
self.seq = seq
self.name = name
self.long_name = long_name
def __str__(self):
return self.seq
def __len__(self):
return len(self.seq)
def Fasta(path):
“”"
Iterator for fasta files
“”"
entry = False
with open(path) as fin:
for line in fin:
if line.startswith(">"):
if entry is not False:
entry.seq = "".join(entry.seq)
yield entry
# define new entry
long_name = line.strip()[1:]
name = long_name.split()[0]
entry = fa_class([], name, long_name)
else:
entry.seq.append(line.strip())
# yield last one
entry.seq = "".join(entry.seq)
yield entry
def gunzip(path, tmp_dir):
“”"
Gunzip a file for EukRep
“”"
if path.endswith(“.gz”):
fna_path = os.path.join(tmp_dir, “contigs.fna”)
logging.debug(“Going to unzip fasta into {}”.format(fna_path))
with gzip.open(path, “r”) as fin, open(fna_path, “w”) as fout:
for line in fin:
fout.write(line.decode())
path = fna_path
logging.debug(“Done unzipping {}”.format(fna_path))
return path
class EukRep:
“”“Class to call and handle EukRep data”“”
def __init__(self, fasta, eukout, bacout=None, minl=1500, tie="euk"):
self.fasta = fasta
self.eukout = eukout
self.bacout = bacout
self.minl = minl
self.tie = tie
def run(self):
# command list will be called
cmd = [
"EukRep",
"--min",
str(self.minl),
"-i",
self.fasta,
"--seq_names",
"-ff",
"--tie",
self.tie,
"-o",
self.eukout,
]
if self.bacout is not None:
cmd.extend(["--prokarya", self.bacout])
subprocess.run(cmd, check=True, shell=False)
self.read_result()
def read_result(self):
self.euks = self.read_eukfile(self.eukout)
self.bacs = set()
if self.bacout is not None:
self.bacs = self.read_eukfile(self.bacout)
def read_eukfile(self, path):
lst = set()
with open(path) as infile:
for line in infile:
lst.add(line.strip())
return lst
class bin:
def init(self, path, eukrep):
self.e = eukrep
self.bname = os.path.basename(path)
self.path = os.path.abspath(path)
def __str__(self):
return "{} euks: {} bacs: {}".format(self.bname, self.table["euks"], self.table["bacs"])
def stats(self):
"""read bin content and figure genomic composition"""
logging.debug("Loading bin")
fa_file = Fasta(self.path)
stats = {"euks": 0, "bacs": 0, "NA": 0, "sum": 0}
# loop and compute stats
最后的话
最近很多小伙伴找我要Linux学习资料,于是我翻箱倒柜,整理了一些优质资源,涵盖视频、电子书、PPT等共享给大家!
资料预览
给大家整理的视频资料:
给大家整理的电子书资料:

如果本文对你有帮助,欢迎点赞、收藏、转发给朋友,让我有持续创作的动力!
logging.debug(“Loading bin”)
fa_file = Fasta(self.path)
stats = {“euks”: 0, “bacs”: 0, “NA”: 0, “sum”: 0}
# loop and compute stats
最后的话
最近很多小伙伴找我要Linux学习资料,于是我翻箱倒柜,整理了一些优质资源,涵盖视频、电子书、PPT等共享给大家!
资料预览
给大家整理的视频资料:
[外链图片转存中…(img-BpnnYp3l-1726129738694)]
给大家整理的电子书资料:
[外链图片转存中…(img-5qwYTXIB-1726129738695)]
如果本文对你有帮助,欢迎点赞、收藏、转发给朋友,让我有持续创作的动力!


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









