









 本文介绍了MMseqs2的安装方法,包括通过conda和静态编译,以及针对Linux系统的性能优化。详细讲解了GTDB_v95数据库的下载与使用,以及如何创建和分析查询数据库,特别是CMake和mmseqs工具在过程中的应用。
本文介绍了MMseqs2的安装方法,包括通过conda和静态编译,以及针对Linux系统的性能优化。详细讲解了GTDB_v95数据库的下载与使用,以及如何创建和分析查询数据库,特别是CMake和mmseqs工具在过程中的应用。
使用方法
github链接:https://github.com/soedinglab/mmseqs2
MMseqs2 User Guide链接: https://mmseqs.com/latest/userguide.pdf
-
安装方式
-
通过conda
conda install -c conda-forge -c bioconda mmseqs2
-
静态安装(AVX2),下载压缩包,解压,将解压后的bin所在的路径添加到环境变量即可
wget https://mmseqs.com/latest/mmseqs-linux-avx2.tar.gz; tar xvfz mmseqs-linux-avx2.tar.gz; export PATH=$(pwd)/mmseqs/bin/:$PATH
-
-
编译(可选)
-
编译可以通过优化特定的系统来提高性能,下面以linux系统为例
git clone https://github.com/soedinglab/MMseqs2.git cd MMseqs2 mkdir build cd build cmake -DCMAKE_BUILD_TYPE=RELEASE -DCMAKE_INSTALL_PREFIX=. .. make make install export PATH=$(pwd)/bin/:$PATH
-
-
数据库下载(GTDB_v95版本)
-
下载链接:https://zenodo.org/record/4751564/files/GTDB_v95.tar.gz?download=1
-
数据库说明:下载的数据库是按照MMseqs2 User Guide的数据库创建方法创建好的,可以直接拿来进行比对。
-
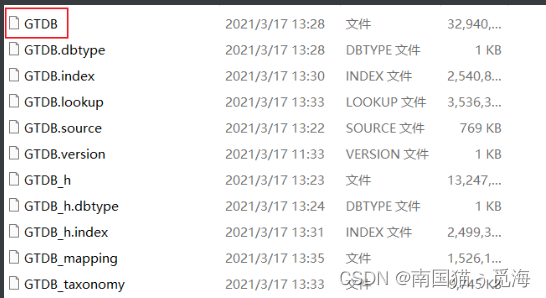
数据库文件如下:(假设红框中的文件位于/data_base/gtdb_db/GTDB路径下)
-
-
创建query数据库(待比对数据集,以CAMI_low数据集为例)
mmseqs createdb /mydata/cami_low.fasta /tmp/query_db # 两个参数,一个是待比对序列,一个是比对数据库
-
注释分类(Taxonomy)
mmseqs taxonomy /tmp/query_db /data_base/gtdb_db/GTDB taxonomyResult tmp # 后面两个参数是必须的,一个是分类结果路径,一个是用于比对过程中存储临时文件的路径,以上命令为默认参数,运行时磁盘空间要在500G以上
-
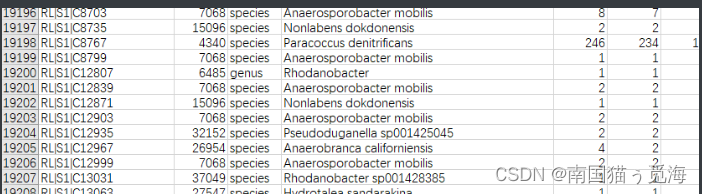
分类结果输出为.tsv格式
mmseqs createtsv /tmp/query_db taxonomyResult taxonomyResult.tsv
-
分类结果如下


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









