







说明:之所以会有数据库制作这一步,是因为按照官网给出的制作命令,运行错误且一直没有找到解决办法。。。
mmseqs databases GTDB gtdb tmp # 官方给出的制作命令,其中GTDB是制作的数据库名称,在mmseqs软件中会有几个常见的数据库,可以直接运行这条命令制作,但名称一定要对,gtdb是制做后的数据库名称,可以自定义,tmp用来存放制作时产生的临时文件。一、准备所需文件
-
https://data.ace.uq.edu.au/public/gtdb/data/releases/latest/bac120_taxonomy.tsv
-
https://data.ace.uq.edu.au/public/gtdb/data/releases/latest/ar53_taxonomy.tsv
将第一个文件gtdb_proteins_aa_reps.tar.gz解压后,包含三个文件:archaea; bacteria; gtdb_release_tk.log
archaea和bacteria文件夹中包含的是待解压的序列,类似于:

首先将上述两个文件夹中的文件解压,然后将它们统一放到一个文件夹(gtdbjieya)下,最后再压缩,并命名为gtdb.tar.gz,解压后的文件如下:
压缩命令:
tar -zcvf gtdb.tar.gz gtdbjieya按照以上步骤得到制作数据库的三个文件:gtdb.tar.gz; bac120_taxonomy.tsv; ar53_taxonomy.tsv
二、根据mmseq提供的databases.sh进行操作
GitHub地址:https://github.com/soedinglab/MMseqs2/blob/master/data/workflow/databases.sh
-
建立一个空文件夹tmp,将以上三个文件放入tmp文件夹下
-
依次运行以下几条命令
1.mmseqs tar2db tmp/gtdb.tar.gz tmp/tardb --tar-include 'faa$' --threads 15
2.sed 's|_protein\.faa||g' tmp/tardb.lookup > tmp/tardb.lookup.tmp
3.mv -f -- tmp/tardb.lookup.tmp tmp/tardb.lookup
4.mmseqs createdb tmp/tardb targetdb/GTDB # targetdb是自己创建的现存的目录,GTDB是数据库的名字(自定义)
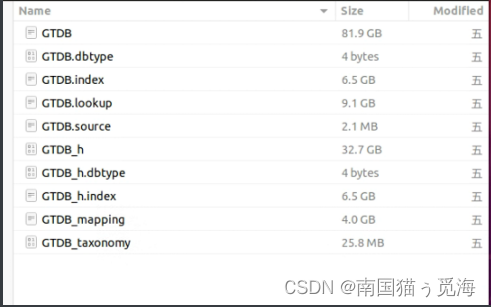
# 上述四条命令运行完,只是完成了"createdb"的操作,会得到以下几个文件:GTDB; GTDB.dbtype; GTDB.index; GTDB.lookup; GTDB.source; GTDB_h; GTDB_h_dbtype; GTDB_h_index; GTDB_taxonomy
# 下面还要完成"createtaxdb"的操作得到一个GTDB_mapping文件
5. CMD='BEGIN {
FS = "[\t;]";
rank["c"] = "class";
rank["d"] = "superkingdom";
rank["f"] = "family";
rank["g"] = "genus";
rank["o"] = "order";
rank["p"] = "phylum";
rank["s"] = "species";
taxCnt = 1;
ids["root"] = 1;
print "1\t|\t1\t|\tno rank\t|\t-\t|" > taxdir"/nodes.dmp";
print "1\t|\troot\t|\t-\t|\tscientific name\t|" > taxdir"/names.dmp";
}
{
prevTaxon=1;
for (i = 2; i <= NF; i++) {
if ($i in ids) {
prevTaxon = ids[$i];
} else {
taxCnt++;
ids[$i] = taxCnt;
r = substr($i, 0, 1);
name = substr($i, 4);
gsub(/_/, " ", name);
printf("%s\t|\t%s\t|\t%s\t|\t-\t|\n", taxCnt, prevTaxon, rank[r]) > taxdir"/nodes.dmp";
printf("%s\t|\t%s\t|\t-\t|\tscientific name\t|\n", taxCnt, name) > taxdir"/names.dmp";
prevTaxon = taxCnt;
}
}
printf("%s\t%s\n", $1, ids[$NF]) > taxdir"/mapping_genomes";
}' # 可以用"$CMD"输出一下,看"CMD"变量是否设置成功
6.mkdir -p tmp/taxonomy
7.awk -v taxdir=tmp/taxonomy "$CMD" tmp/bac120_taxonomy.tsv tmp/ar53_taxonomy.tsv
8.touch tmp/taxonomy/merged.dmp
9.touch tmp/taxonomy/delodes.dmp
10.mmseqs createtaxdb targetdb/GTDB tmp/taxdb --ncbi-tax-dump tmp/taxonomy --tax-mapping-file tmp/taxonomy/mapping_genomes --tax-mapping-mode 1 --threads 15
# 上述命令运行完你会得到一个GTDB_mapping文件,至此数据库创建完毕。数据库文件如下(在targetdb目录下,tmp中的文件可自行删除):

















 2056
2056

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









