






接shan老师@shanshandeisu的分子动力学模拟文章最后的mmpbsa部分:网络药理学之薛定谔Schrödinge Maestro:8、分子对接和分子动力学模拟速通版总结(前置处理、后续分析和可视化)_薛定谔如何显示对接结果-CSDN博客
使用wsl子系统的玩家可以从头开始看,组内同学可以直接跳到运行MMPBSA部分
MMPBSA脚本获取
mmpbsa用的是李继存老师的脚本,网址:https://github.com/Jerkwin/gmxtools/tree/master/gmx_mmpbsa
除了需要MMPBSA脚本外,还需要安装gromcas(相信大家都有)以及安装apbs,可以通过一下命令安装:sudo apt install apbs。组内的同学已经在服务器中配备好了环境,不过感兴趣可以看一下。
gmx_mmpbsa.bsh部分参数解读,原代码如下:
trj=traj.xtc # 轨迹文件 trajectory file
tpr=topol.tpr # tpr文件 tpr file
ndx=index.ndx # 索引文件 index file
pro=Protein # 蛋白索引组 index group name of protein
lig=Ligand # 配体索引组 index group name of ligand
step=0 # 从第几步开始运行 step number to run
# 1. 预处理轨迹: 复合物完整化, 团簇化, 居中叠合, 然后生成pdb文件
# 2. 获取每个原子的电荷, 半径, LJ参数, 然后生成qrv文件
# 3. MM-PBSA计算: pdb->pqr, 输出apbs, 计算MM, APBS
# 1. pre-processe trajectory, whole, cluster, center, fit, then generate pdb file
# 2. abstract atomic parameters, charge, radius, C6/C12, then generate qrv file
# 3. run MM-PBSA, pdb->pqr, apbs, then calculate MM, PB, SA
gmx='gmx' # /path/to/GMX/bin/gmx_mpi
dump="$gmx dump" # gmx dump
trjconv="$gmx trjconv -dt 1000" # gmx trjconv, use -b -e -dt, NOT -skip
converttpr="$gmx convert-tpr" # to generate reduced tpr
#apbs='/path/APBS/bin/apbs' # APBS(Linux)
apbs='E:/APBS1.5/apbs.exe' # APBS(Windows), USE "/", NOT "\"
export MCSH_HOME=/dev/null # APBS io.mc
这里可以把轨迹文件、拓扑文件、索引文件、蛋白索引和配体索引改为自己的,同时更改apbs的路径,以及删去export MCSH_HOME=/dev/null,这段代码的作用是将apbs产生的临时文件丢入 Linux 的空设备文件 /dev/null,由于用的是wsl子系统,就不用这条命令。
运行MMPBSA
首先是激活环境,在完成分子对接后,记得关闭当前窗口,重开一个窗口,通过以下命令:
# 环境激活
module load miniforge3/24.11
source activate MMPBSA
module load gromacs/2022.5
cd run/ # 这里cd到你自己的路径随后就可以进行正式的MMPBSA脚本运行,首先是轨迹采样,选择一段比较平稳的时间段,比如我这里选择的是50-70ns,进行轨迹采样:
gmx trjconv -f md_nojump.xtc -o traj.xtc -b 50000 -e 70000
gmx check -f traj.xtc # 检查轨迹 -b 50000指的是从50000ps(50ns)开始采样,-e 70000指的是从70000(70ns)停止。运行后我们需要从服务器中下载三个文件:
1、index.ndx:索引文件
2、traj.xtc:轨迹文件
3、md.tpr:拓扑文件
将准备文件移动到工作目录后,输入以下命令即可运行脚本:
bash gmx_mmpbsa.bsh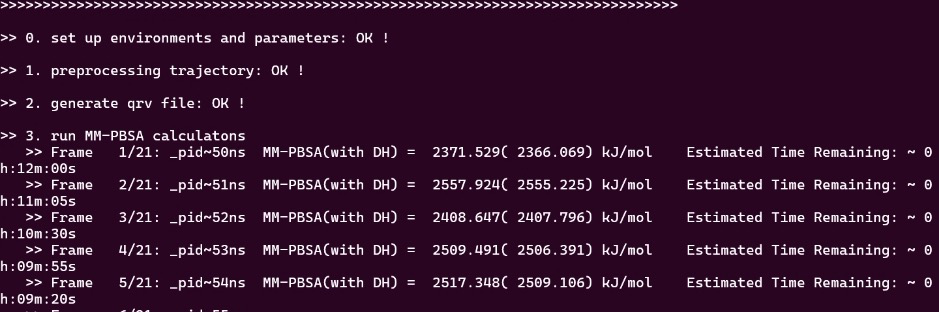
可以看到运行时间是比较久的,博主cpu是ultra7 265k,满载需要运行12分钟,没有调用显卡,各位在运行脚本的时候可以以此为参考分配时间。
数据后处理
MMPBSA数据处理的核心就是把结合能和氨基酸残基意义对应,这里以运行结果中_pid~res.dat处理为例子,展示如何处理数据。将其中的数据复制到一个表格中
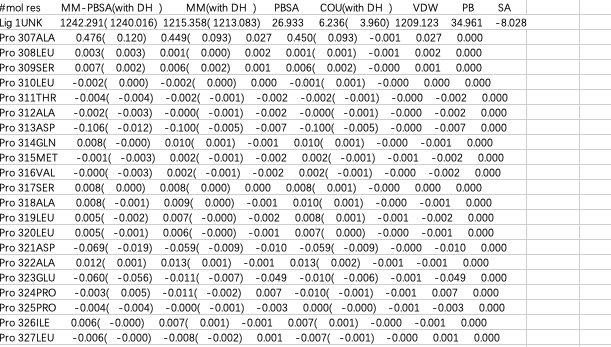
随后选中数据-分列-固定宽度,再删去A、D、F、I列(删去with DH),再使用替换删掉所有的括号,这样就可以把想要研究的两列结合能量选中,复制到一个txt中,并在表头添加:
@ title "MMPBSA"
@ xaxis label "Protein"
@ yaxis label "Energy (kj/mol)"
@TYPE xy
下面我做一个演示,首先,整理后到表格内容如下:
如果我想要研究MMPBSA分解到各个残集的图,我可以选中A、B两列,复制到一个txt中,删去第一、第二列得到:
随后将上面提到的这段带@的文字复制到最开头
保存后将txt后缀更改为.xvg,再拖到qtgrace中即可。
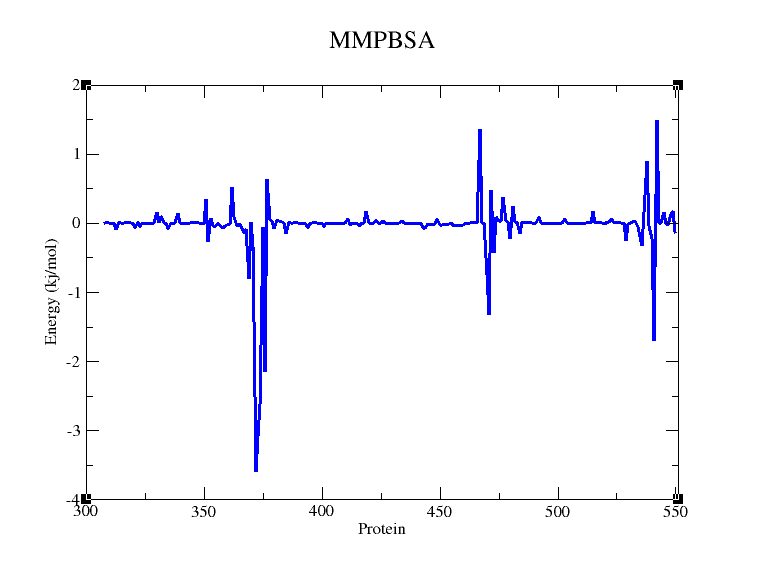
最后调整一下颜色和线条的粗细(也可以标一下氨基酸,博主这边就不标了,只做演示),就可以得到想要的图:


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









