






还想纯生信发文?这样做就对了!
很多人说,现在纯生信发文越来越难,竞争激烈、审稿严格,简直“内卷”到极致!但真的就无路可走了吗?其实不然! 纯生信发文难,但并非不可行,只要走对路,你也能在学术的森林中杀出一条光明大道!
关键在于两点:拼选题和拼思路!
1️⃣ 拼选题:选题即战场,好的选题是成功的一半!
找一个学术热点,切入一个创新角度,或者聚焦未被充分研究的小众领域,都能让你的研究脱颖而出。尤其是交叉学科领域,比如结合肿瘤学、免疫学等热门方向,或者挖掘特定人群或罕见病的独特机制,这些都可能成为你的加分项。
2️⃣ 拼思路:有创意的分析思路,才是你的核心竞争力!
生信研究的价值不仅在于“发现了什么”,更在于“怎么发现”。与其追求大而全,不如深挖一个问题,从数据预处理到分析方法都力求精细,甚至尝试结合机器学习或多组学整合等新技术,展示你的独特视角和专业功底。
只要选对方向、方法得当,纯生信研究一样可以“卷”出新高度! 所以,不要被困难吓倒,试着从选题和思路上下功夫,你会发现,发文不再是遥不可及的梦!
快来留言分享你的选题灵感和研究思路吧,说不定我们能一起擦出学术的火花!
今天,我们来分享一篇“拼思路”的经典案例:纯生信发到了2区、6.1分的生信友好宝藏刊物!✨
这篇文章的选题方向是线粒体——常见;研究对象是肺腺癌——大癌种,也常见。那它凭什么脱颖而出?靠的就是联合思路!单细胞分析 + 机器学习 + Mendelian随机化分析,一篇文章用上了这些热门分析方法,整合起来效果奇佳。

题目:综合多组学整合揭示线粒体基因特征在肺腺癌预后与个性化治疗中的应用价值
杂志:Journal of Translational Medicine
影响因子:IF=6.1
中科院分区:医学二区
发表时间:2024年10月
PART·1 研究背景
肺癌(LC)是全球致死率最高的癌症之一,2020年约180万人因此死亡。肺腺癌(LUAD)作为非小细胞肺癌(NSCLC)的主要亚型,占肺癌病例的40%,且比例持续上升。尽管多模式治疗有所进展,LUAD患者的5年生存率仍偏低。深入研究其分子机制,寻找可靠生物标志物和新治疗靶点,对改善预后和实现个性化治疗至关重要。
PART·2 方法学
数据下载与预处理
本研究从The Cancer Genome Atlas (TCGA)数据库(https://portal.gdc.cancer.gov)获取了肺腺癌(LUAD)全转录组数据及临床信息,共包含513个样本。同时,从Gene Expression Omnibus (GEO)数据库(http://www.ncbi.nlm.nih.gov/geo)下载了转录组和单细胞数据。
转录组数据集包括:GSE81089(n=107)、GSE13213(n=117)、GSE14814(n=71)、GSE26939(n=115)、GSE31210(n=226)、GSE30219(n=85)、GSE37745(n=106)、GSE50081(n=127)、GSE42127(n=133)、GSE72094(n=398)和GSE68465(n=442)。
单细胞数据集包括:GSE131907(包含11个邻近非肿瘤样本、11个肿瘤样本及4个IV期肿瘤样本)和GSE127465(18个肿瘤样本),总计44个样本。
 图1.研究流程图
图1.研究流程图
单细胞测序分析
本研究使用Seurat R包对GSE131907数据集的单细胞数据进行聚类、降维和亚分类分析。主要目标是通过t-SNE图直观展示细胞群及其分布。利用“FindAllMarkers”函数识别不同细胞群中的差异表达基因,并将结果可视化为热图,同时将这些基因作为细胞类型注释的标记基因。
成纤维细胞、T细胞及上皮细胞的转录活性分析
使用“decoupleR”软件结合Dorothea对不同细胞亚群的转录因子(TF)进行分析。在细胞水平计算活性得分,以描述每个细胞中转录因子及其下游靶基因(调控子)的富集情况。
GSEA分析与功能注释
为了评估上皮细胞亚群和肿瘤亚型的功能特征,使用“clusterProfiler”包对上皮细胞亚群和肿瘤亚型中差异上调的基因进行基因集富集分析(GSEA)。富集分析基于分子特征数据库(MSigDB)3.0的Hallmark基因集进行。当Benjamini & Hochberg (BH)校正后的p值<0.05时,通过“enrichplot”包对富集结果进行可视化展示。
基于机器学习集成方法构建的风险模型
为了开发一个高精度和稳定性的预后模型,研究团队集成了10种机器学习算法及其101种组合,构建了AIDPS模型。集成算法包括弹性网(Elastic Net, Enet)、随机生存森林(Random Survival Forest, RSF)、岭回归(Ridge)、逐步Cox回归(Stepwise Cox)、最小绝对收缩和选择算子(LASSO)、CoxBoost、监督主成分分析(SuperPC)、广义提升回归模型(GBM)、生存支持向量机(survival-SVM)和Cox偏最小二乘回归(plsRcox)。
AIDPS模型评估
为了验证AIDPS模型的预测性能和稳健性,对12个数据集和合并样本的元队列进行了接收操作特征(ROC)分析,通过条形图可视化不同时间点(1年、3年和5年)的曲线下面积(AUC)值。对于TCGA-LUAD队列和11个验证集,计算了AIDPS模型和多个临床指标的C指数值,并生成误差条形图,直观比较不同C指数值的差异。
化疗和免疫治疗预测反应分析
为了评估TCGA-LUAD队列中不同组别对化疗反应的差异,研究团队基于“oncoPredict”包计算了几种传统化疗药物的半最大抑制浓度(IC50)。
肿瘤SNV和免疫微环境分析
从TCGA数据库中获取了SNV数据,详细分析了SNV突变类型、基因突变率以及不同组别之间的突变差异,并通过瀑布图展示了结果。随后,使用“maftools”包计算了不同组别的肿瘤突变负荷(TMB)、同源重组缺陷(HRD)和突变等位基因肿瘤异质性(MATH)值。
LC的GWAS数据分析和MR分析
LC的GWAS数据来源于全基因组复杂性状分析(GCTA)网站,并使用“CMplot”包绘制了GWAS的曼哈顿图。同时,使用SMR软件,以欧洲人群的千人基因组数据为参考,进行基因共定位分析,基于eQTLGen网站(https://eqtlgen.org/cis-eqtls.html)的cis-eQTL数据和LC-GWAS数据进行分析。随后,使用R中的“TwoSampleMR”包进行MR分析,重点探讨与预后模型相关的基因与单核苷酸多态性(SNP)位点之间的关联,以及间质性肺病(ebi-a-GCST90018643)与LC之间的关联。分析结果通过散点图、森林图和漏斗图进行可视化展示。
统计分析
数据处理、统计分析和可视化使用R软件(版本4.1.3)进行。皮尔逊相关系数用于评估两个连续变量之间的相关性。卡方检验用于比较分类变量,威尔科克森秩和检验或T检验用于比较连续变量。使用“survminer”包确定最佳截断值。Cox回归和Kaplan-Meier分析使用“survival”包进行。“CompareC”包用于比较不同变量的C指数值。图表使用“ggplot2”包生成,p值小于0.05被认为具有统计学意义(* p < 0.05;** p < 0.01;*** p < 0.001;**** p < 0.0001)。
PART·3 分析结果
肺腺癌(LUAD)单细胞表达图谱
通过对GSE131907数据集进行单细胞分析,识别出31个细胞群,并通过t-SNE图展示正常和肿瘤组织细胞的分布差异。基于31个标记基因,将细胞注释为八种类型,并通过气泡图验证分类准确性。肿瘤组织中T细胞和B细胞比例较高,正常组织中内皮细胞、NK细胞和髓系细胞比例较高。
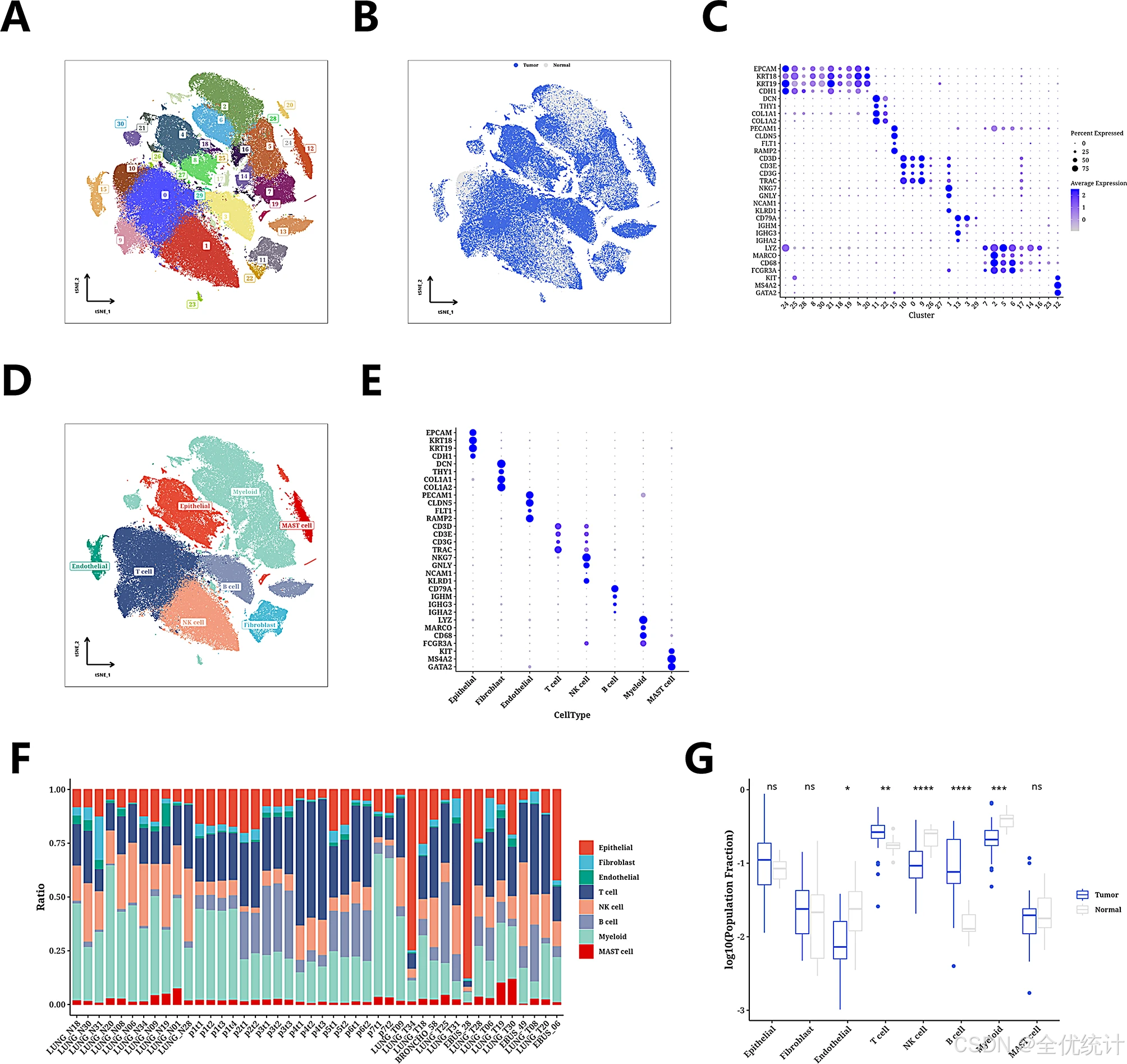 图2.单细胞表达图谱
图2.单细胞表达图谱
构建和验证PRGs
随后,研究团队分离了上皮细胞和T细胞进行独立的降维和聚类分析。t-SNE图揭示了14个不同的群体,表明大多数T细胞来源于肿瘤样本。
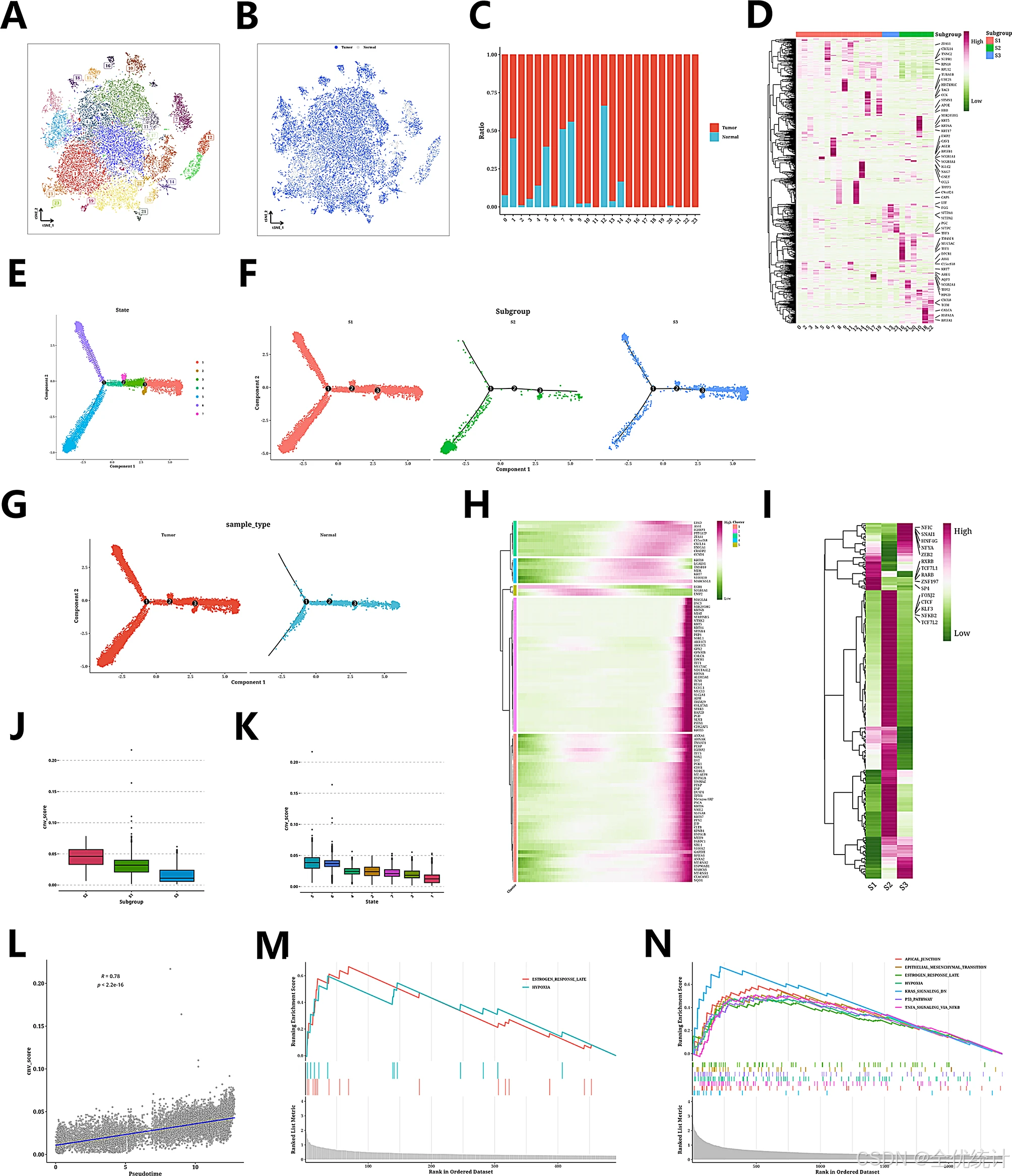 图3.上皮细胞的亚类分析、细胞轨迹分析和CNV分析结果
图3.上皮细胞的亚类分析、细胞轨迹分析和CNV分析结果
 图4.T细胞和CAF细胞的的亚分类分析
图4.T细胞和CAF细胞的的亚分类分析
通过机器学习集成方法构建的预后风险模型
在TCGA-LUAD数据集中,使用LOOCV框架拟合101个预测模型,并计算每个模型在11个验证数据集中的C指数。最佳模型Enet[a = 0.2](平均C指数0.655)在所有验证数据集中表现最佳。根据预后基因表达计算患者风险评分,并按临界值分为高风险组和低风险组。在TCGA-LUAD及其他11个验证数据集中,高风险组的总体生存期显著低于低风险组(p < 0.05)。在所有样本的元队列中也观察到了相似趋势(p < 0.05)。
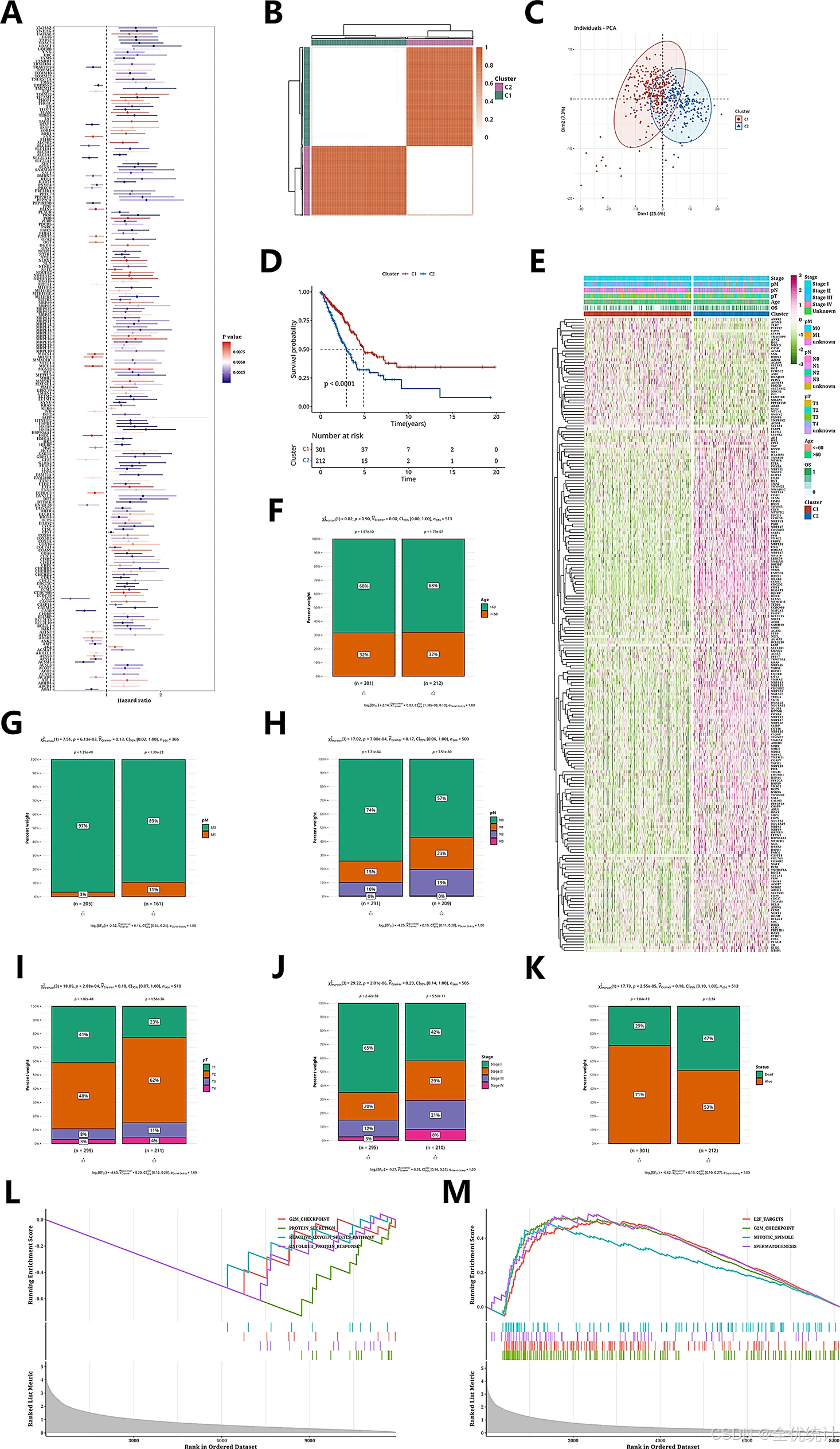 图5.线粒体相关预后基因的鉴定和聚类分析
图5.线粒体相关预后基因的鉴定和聚类分析
AIDPS模型评估
过ROC分析评估了AIDPS在12个数据集及元队列中的预测性能。在TCGA-LUAD中,1年、3年和5年生存的AUC分别为0.682、0.695和0.657。其他数据集的AUC值也显示出良好的预测能力。一致性指数(C-index)误差条形图显示大多数数据集的C-index值超过0.6,证明AIDPS在不同队列中的稳定性和可靠性。此外,AIDPS相比传统临床特征(如性别、年龄、病理分期和吸烟状态)在预后预测中的准确性更高。
利用ROC曲线评估AIDPS模型的预测性能,AUC值突出了AIDPS的稳健预测能力。此外,比较AIDPS与传统临床特征的预测能力,结果显示,AIDPS的准确性优于性别、年龄、病理阶段和吸烟状况等因素。
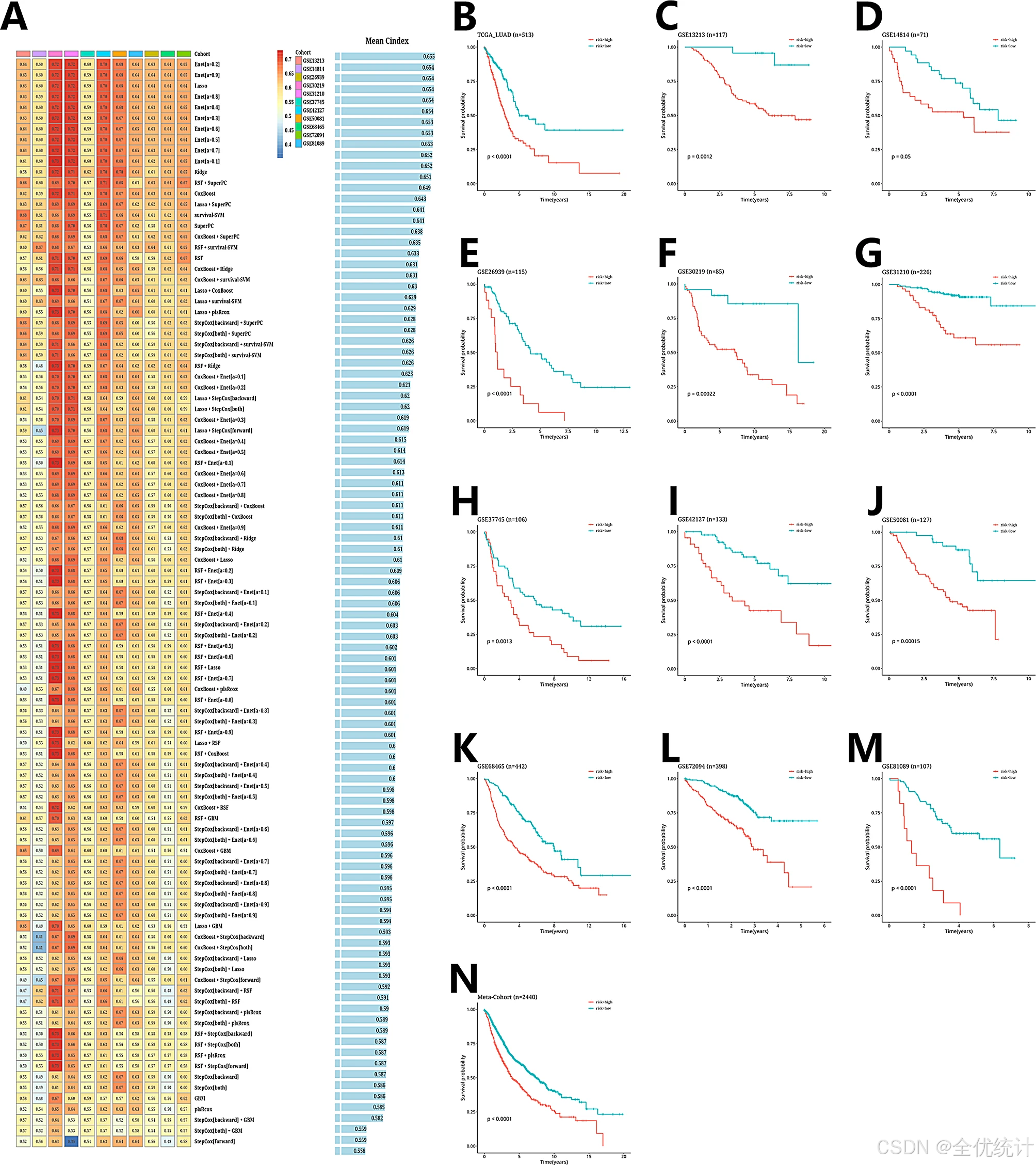 图6.预后模型构建
图6.预后模型构建
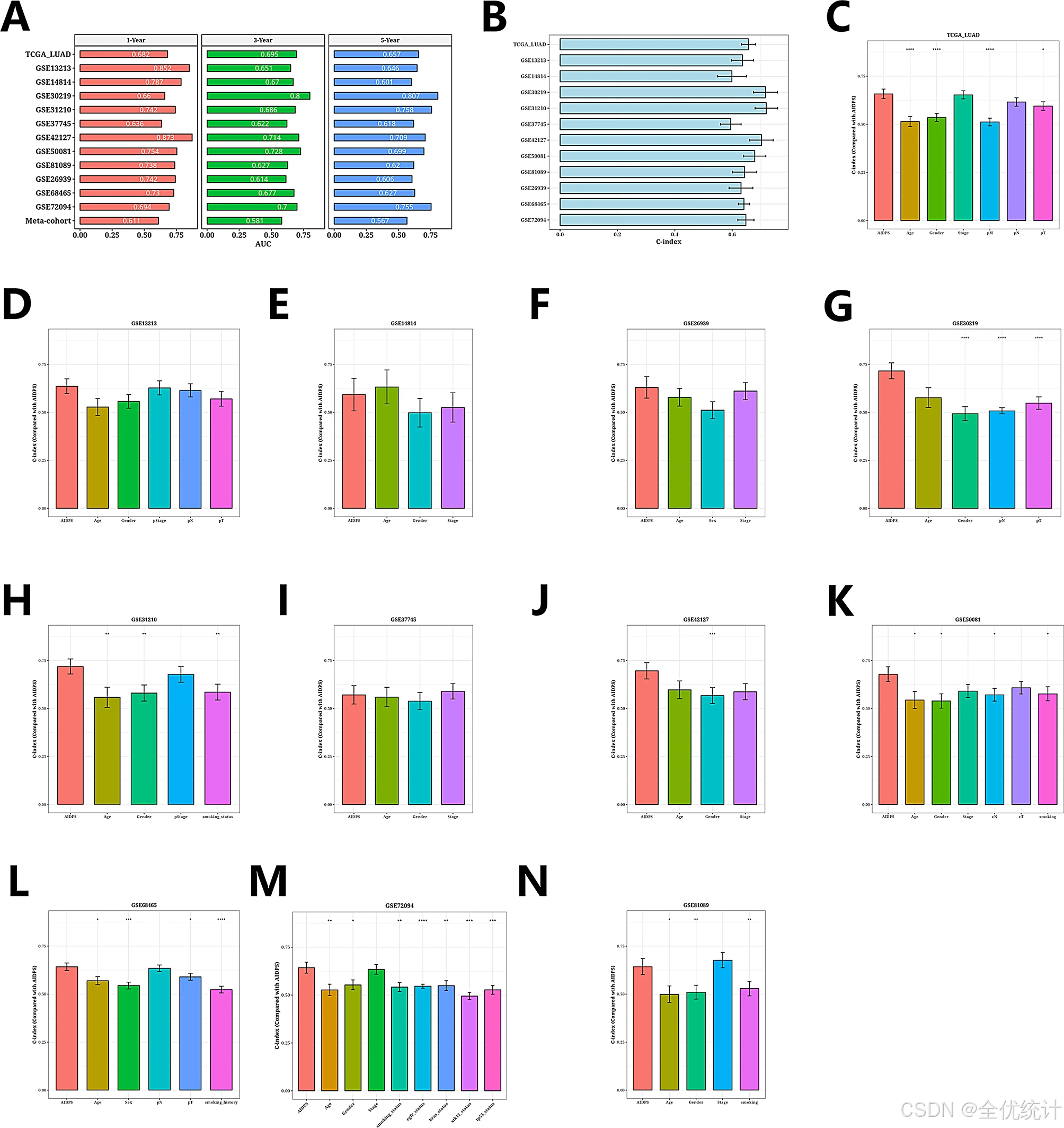 图7.AIDPS模型评价
图7.AIDPS模型评价
高低风险组对化疗和免疫治疗预测反应的分析
研究团队发现化疗药物Bortezomib_1191、Docetaxel_1007、Sepantronium bromide_1941和Vinblastine_1004的半数抑制浓度(IC50)在高低风险组之间存在显著差异,其中高风险组的IC50值始终显著低于低风险组(p < 0.05)。此外,利用TIDE(肿瘤免疫功能障碍与排除)算法分析显示,高风险组的TIDE评分显著高于低风险组(p < 0.05)。生存分析表明,高风险无应答组的预后显著差于高风险应答组、低风险无应答组和低风险应答组(p < 0.0001)。
 图8.与其他基于文献的预后模型的比较
图8.与其他基于文献的预后模型的比较
肿瘤SNVs及免疫微环境分析
瀑布图展示了两个风险组中突变频率最高的前30个基因的突变状态,其中TP53(48.6%)、TTN(43.2%)和MUC16(39.4%)的突变频率相对较高。
 图9.风险组对免疫治疗/化疗反应预测的分析结果
图9.风险组对免疫治疗/化疗反应预测的分析结果
肺癌的GWAS数据分析及MR分析
对肺癌GWAS数据的曼哈顿图分析显示,在22条染色体上存在多个显著的SNP位点,其中最显著的位点位于染色体2(p < 10⁻⁴)。随后,利用eQTLGen和LC-GWAS数据通过SMR软件进行基因共定位分析,鉴定出两个与预后相关的基因:CDKN3和MYO1E。在用于预后模型构建的Cox基因相关SNP位点的孟德尔随机化(MR)分析中,未发现间质性肺病(ebi-a-GCST90018643)与肺癌(ukb-a-54)之间的显著关联。然而,某些SNP位点(如rs1794002和rs244320)在这两种疾病中均表现出显著关联。
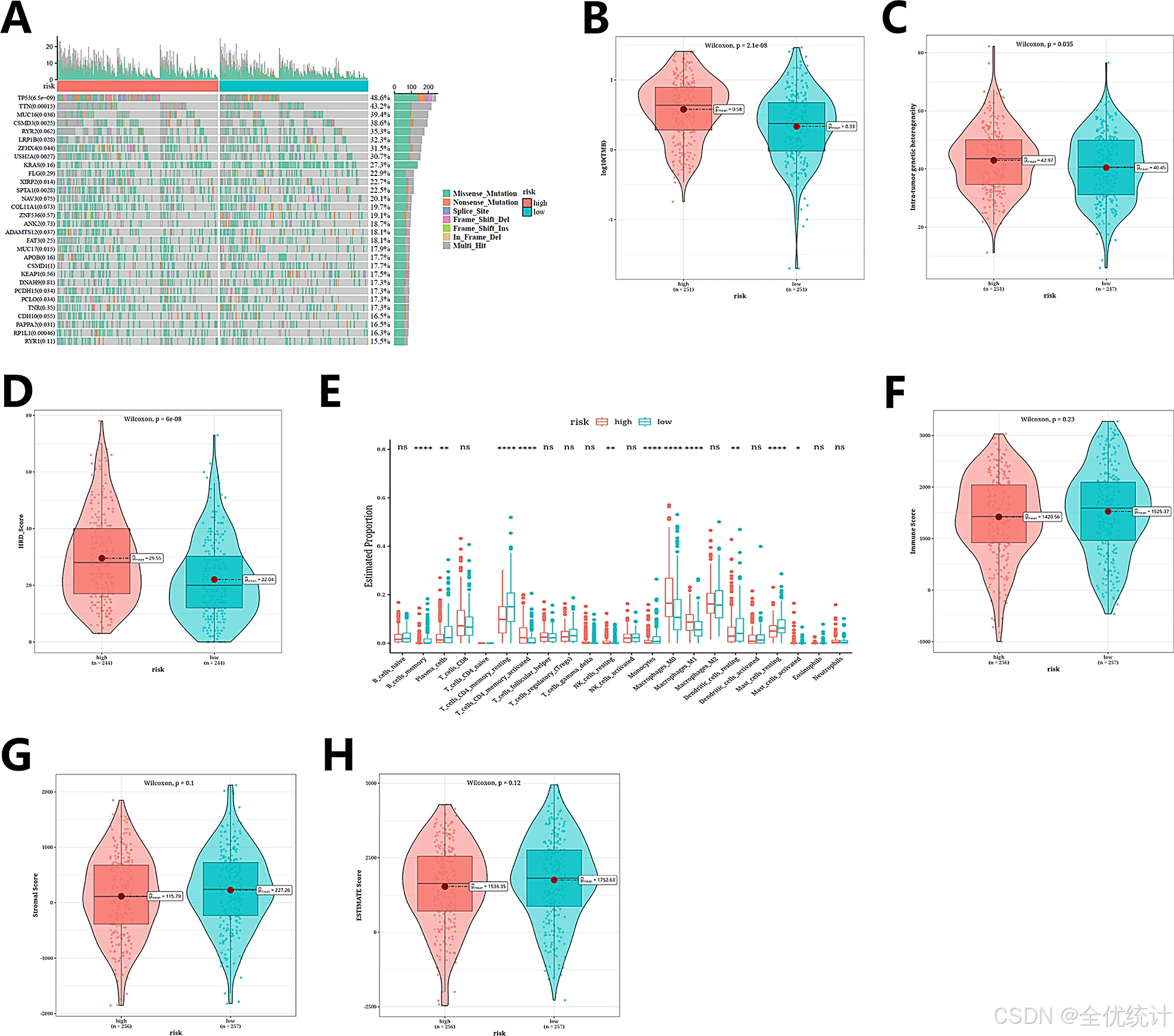 图10.SNV突变和免疫微环境差异分析结果
图10.SNV突变和免疫微环境差异分析结果
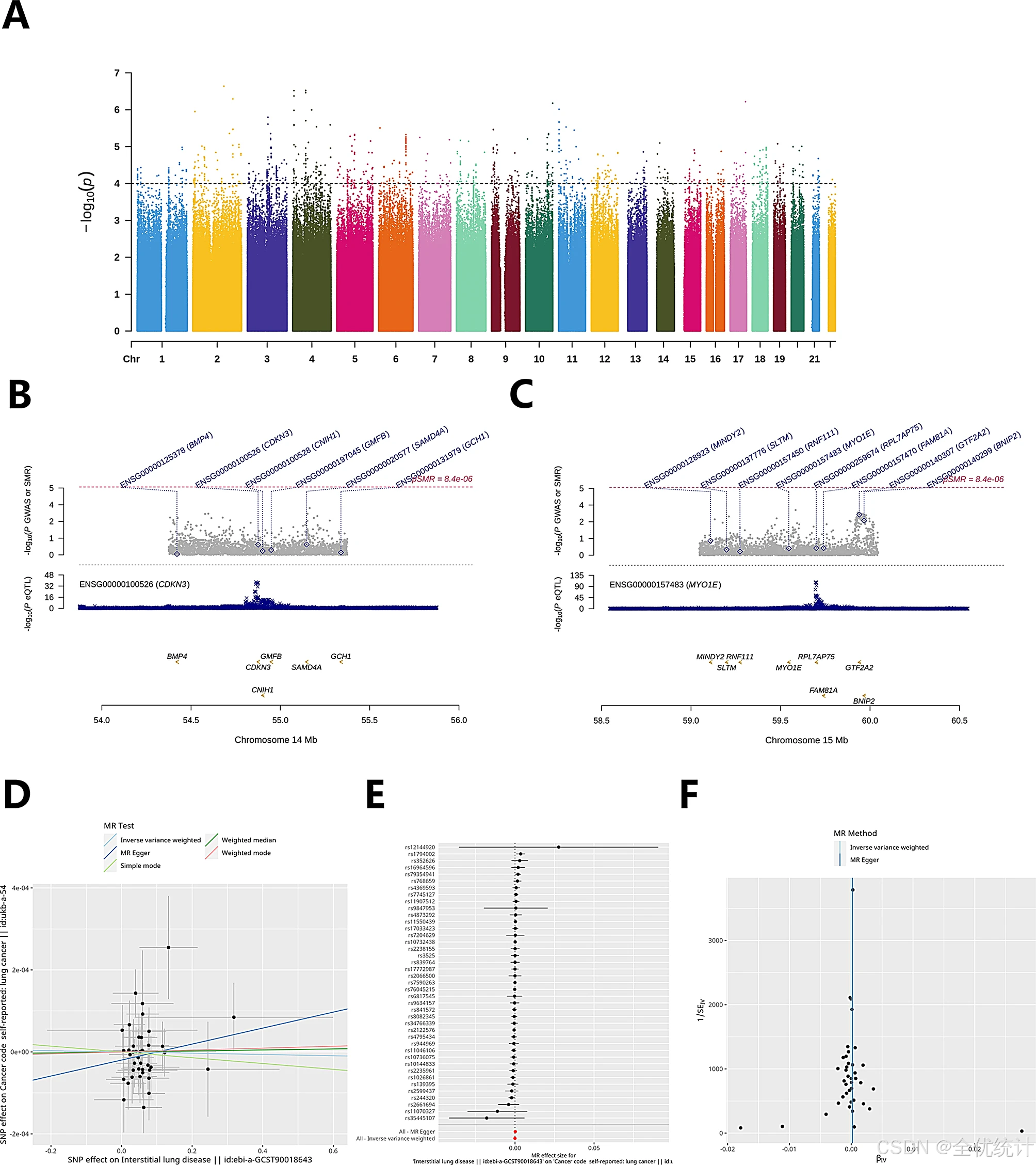 图11.遗传关联和MR分析结果
图11.遗传关联和MR分析结果
PART·4 小优总结
在这项研究中,研究团队深入分析了肺腺癌(LUAD),采用了多种方法,包括获取和处理转录组数据和单细胞RNA测序(scRNA-seq)数据,并进行了细胞分类与亚分类分析。此外,还开展了免疫浸润特征分析、免疫治疗和化疗反应预测、基因集富集分析(GSEA)以及单核苷酸变异(SNV)分析。这些生物信息学方法的综合应用建立了一个全面且多维的数据集,为深入理解LUAD的分子特征和免疫动态提供了重要的视角。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









