



粪肠球菌fsr群体感应回路强效激活剂和抑制剂的合理设计
摘要
细菌对抗生素耐药性的日益增长,迫切需要寻找替代治疗方案,以降低细菌的适应能力。粪肠球菌仍然是临床肠球菌感染的主要致病菌,并表现出依赖群体感应(QS)的致病性。本文报道了针对粪肠球菌Fsr群体感应系统的基于大环肽的激活剂和抑制剂的开发。为此,我们开发、优化并比较了三种含内酯的大环肽骨架的合成路线。随后,结合先前及当前对天然群体感应信号肽的构效关系(SAR)研究,理性设计出迄今为止最强效的Fsr群体感应系统激活剂和抑制剂。这些肽的应用有望在不显著增加细菌产生耐药性的选择压力的前提下,减弱粪肠球菌的致病性。
引言
粪肠球菌是一种共生的革兰氏阳性细菌,也是一种机会性病原体,尽管屎肠球菌感染呈上升趋势,但粪肠球菌仍是临床肠球菌感染的主要原因。粪肠球菌通常存在于人体肠道中,但可引起血液感染(菌血症)、尿路感染以及心脏定植(心内膜炎)。粪肠球菌对多种抗生素具有固有耐药性,并能快速产生或获得新的抗生素耐药性。此外,研究表明粪肠球菌能够与其他物种共享耐药基因,包括多重耐药菌株如耐甲氧西林金黄色葡萄球菌(MRSA)。临床粪肠球菌感染带来的医疗负担及其发展和传播抗生素耐药性的能力,凸显了开发替代策略以根除粪肠球菌感染的必要性。
一种对抗细菌感染的传统抗生素替代策略是开发能够减弱细菌致病性而非诱导细胞死亡的分子。这种方法在减少选择压力以防止耐药性发展的同时,仍能消除许多有害的细菌的影响。粪肠球菌是该方法的一个理想靶点,因为它表现出对群体感应(QS)的依赖性,以协调致病性群体行为,例如生物膜形成和毒力因子产生。
群体感应是细菌用来评估细胞密度并协调群体行为(包括生物膜形成、毒力因子产生、孢子形成、感受态诱导、群集运动和生物发光)的一种通讯方式。革兰氏阳性细菌中的群体感应依赖于自诱导肽(AIPs),这些肽由细菌产生并分泌。细菌利用AIPs来感知局部细胞密度,并负责激活群体感应通路。当AIP浓度达到足够高时,会激活一种膜结合受体(通常为组氨酸激酶受体),从而导致群体感应基因的上调(以同步细胞,使其进入群体行为状态)以及激活群体行为的相关基因。
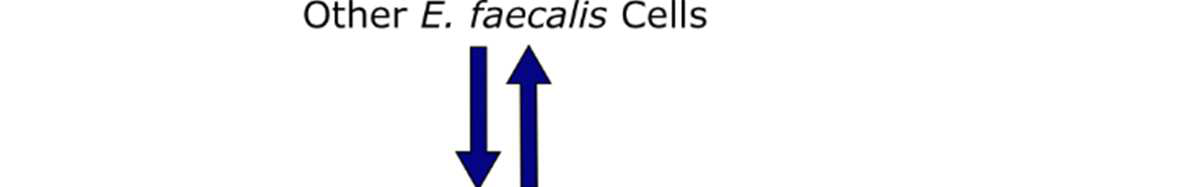
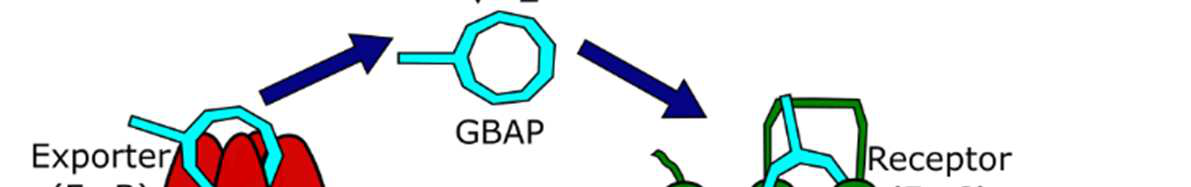
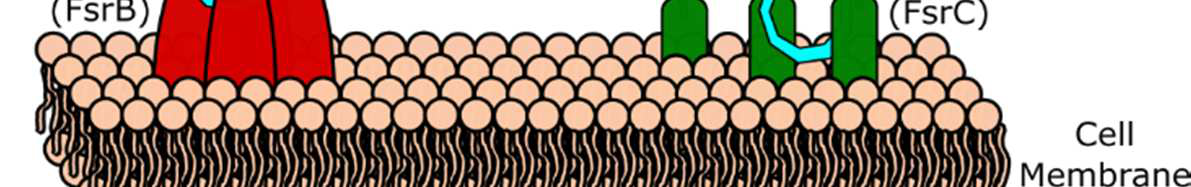
在粪肠球菌中,致病性的主要调控系统称为Fsr群体感应系统(图1)。该系统利用一种含有11个氨基酸的环状自诱导肽,称为明胶酶生物合成激活信息素(GBAP)以激活Fsr依赖性基因。该肽具有一段从肽N端延伸出的双氨基酸尾部。该尾部从一个丝氨酸残基延伸而出,该残基对于其侧链与C端甲硫氨酸残基的羧酸之间形成内酯大环连接至关重要。本课题组及其他团队开展的构效关系(SAR)研究已确定了关键的侧链及其手性取向,以及可耐受修饰的位点。此外,已有研究评估了阻断Fsr群体感应系统的治疗潜力,并开发出一种基于GBAP的强效竞争性抑制剂,能够减弱粪肠球菌的群体感应依赖性致病性。
本研究中,我们报道了基于GBAP的Fsr群体感应系统的激动剂和拮抗剂的合理设计。为此,我们利用先前报道的构效关系信息,提出作用机制假设,并在构建非生物GBAP类似物过程中加以实施。该工作扩展了对GBAP分子的构效关系知识,获得了迄今为止活性最强且活性处于低至亚纳摩尔范围的激动剂和拮抗剂。

结果与讨论
疏水环区(立体)构效关系研究
为了进一步明确GBAP(1)环区的构效关系,特别是Phe7、Gly8和Trp10在受体结合与激活中的作用,我们通过引入保守点突变来评估这些位点。首先,我们发现将Gly8替换为丙氨酸(GBAP-G8A,2)导致完全丧失活性。由于该修饰实际上仅引入了一个手性中心,我们推测Gly8的作用是使肽链在此位置能够形成更类似于D-氨基酸的构象。因此,用D-丙氨酸取代Gly8应更具耐受性,并产生能够有效结合并激活FsrC受体的类似物。事实上,将Gly8替换为D-丙氨酸(GBAP-G8a,3)相较于天然GBAP仅表现出轻微的效价降低(效价降低20倍,而在此位置引入L-丙氨酸则完全丧失活性;表1)。
之前,我们发现Phe7位置的手性翻转(GBAP-f7,4)出人意料地被很好地耐受(效价仅降低5倍)。该结果进一步支持了我们的假设,即该区域需要类似D-氨基酸的取向(由天然GBAP中甘氨酸提供的灵活性实现),以使GBAP呈现生物活性构象。此外,尽管这一结果可能已经部分可归因于相邻甘氨酸的灵活性,也有可能是受体结合口袋中的空间混杂性允许该位置不同侧链及侧链取向的有效结合。为了验证第二种可能性,制备了Phe7被色氨酸和D-色氨酸取代的类似物。结果发现,色氨酸取代(F7W,5和F7w,6)比相应的苯丙氨酸类似物效价略高(表1)。然而,将天然的Trp10和Phe7残基互换得到7(GBAP-F7WW10F),其效价降低了27倍。此外,将Trp10替换为Phe(GBAP-W10F,8)得到的类似物效价降低了5倍。最后,将两个残基均替换为丙氨酸(GBAP-F7AW10A,9)导致活性完全丧失,这一结果与先前报道一致。综合来看,这些结果强调了第10位较大体积的色氨酸侧链的重要性,而Phe7残基则具有可修饰性,可能通过优化以增强肽-受体结合。此外,7号化合物相较于两个单取代类似物(5和8)效价显著降低,表明该残基交换引起了相对于天然GBAP或单取代类似物的显著构象变化。最后,已证实第7位具有空间混杂性,表明Phe残基在FsrC中并未完全占据其结合口袋。然而,该位置可接受修饰的范围尚未完全确定。
尾部和环化构效关系研究
我们先前报道过,乙酰基(图2A)作为足够的环外侧链足以维持GBAP的活性(10,表1)。相反,一个缺乏该侧链、通过内酰胺环(头尾环化)进行环化的类似物则完全无活性(11),而在丝氨酸3(Ser3)引入D-氨基酸突变的GBAP-s3(12)则导致活性降低40倍。综合来看,这些结果表明Ser3的手性反转引起了构象变化,导致环外侧链与FsrC结合位点发生空间冲突。因此,我们假设在此类似物中将侧链替换为乙酰基将提高其活性。事实上,Ac-GBAP-Des(Q1N2)s3(13)相较于全长类似物(12)的效价提高了6倍。有趣的是,一种内酰胺头尾环化类似物(14)的活性也被发现高于其L-丝氨酸3对应物(11),这表明即使侧链未直接参与环化,丝氨酸3中构型的反转也会引起环构象的显著变化。

此前发现,GBAP容易发生尾部降解,在N端形成焦谷氨酸环(图2B)。我们推测GBAP固有的不稳定性可能具有调控作用,使细菌能够准确感知其种群密度的变化。为了验证这一假设,我们合成了焦谷氨酸降解产物的D型和L型,并对其进行了活性评价。结果发现,L型(15)的活性与天然GBAP相当,而D型(16)的活性则低约3倍。这些结果否定了焦谷氨酸降解是因进化压力被选择出来并在评估种群密度中发挥调控作用的观点。
表1. 疏水环区保守取代、尾部和环化类似物的EC50值
| 肽# | 肽名称 | 序列 | EC50 [95% CI] (nM) | Fold变化 |
|---|---|---|---|---|
| 1 | GBAP | QN(SPNIFGQWM) | 1.15[0.825–1.59] | NA |
| 2 | GBAP-G8A | QN(SPNIFAQWM) | >10,000 | (>10,000) |
| 3 | GBAP-G8a | QN(SPNIFaQWM) | 20.8[8.65–50.1] | (18) |
| 4 | GBAP-f7 | QN(SPNIfGQWM) | 4.97[3.12−7.77] | (4.3) |
| 5 | GBAP-F7W | QN(SPNIWGQWM) | 0.905[0.625–1.31] | 1.3 |
| 6 | GBAP-F7w | WN(SPNIwGQWM) | 1.82[0.710–4.64] | (1.6) |
| 7 | GBAP-F7WW10F | QN(SPNIWGQFM) | 27.4[17.7–42.4] | (24) |
| 8 | GBAP-W10F | QN(SPNIFGQFM) | 4.86[3.43–6.89] | (4.2) |
| 9 | GBAP-F7AW10A | QN(SPNIAGQAM) | >10,000 | (>10,000) |
| 10 | Ac-GBAP-Des(Q1N2) | (SPNIFGQWM) | 1.01[0.496–2.06] | 1.1 |
| 11 | GBAP-Des(Q1N2) lactam | (SPNIFGQWM) | >10,000 | (>10,000) |
| 12 | GBAP-s3 | QN(sPNIFGQWM) | 45.1[17.0–120] | (39) |
| 13 | Ac-GBAP-Des(Q1N2)s3 | Ac-(sPNIFGQWM) | 7.58[2.95–19.5] | (6.6) |
| 14 | GBAP-Des(Q1N2)s3 内酰胺 | (sPNIFGQWM) | 764[446–1311] | (664) |
| 15 | GBAP-Q1[焦谷氨酸] | [PyGlu]-N(SPNIFGQWM) | 1.14[0.818–1.59] | 1.0 |
| 16 | GBAP-q1[焦谷氨酸] | [D-PyGlu]-N(SPNIFGQWM) | 3.11[2.05–4.72] | (2.7) |
| 17 | Z-GBAP | Z-QN(SPNIFGQWM) | 0.178[0.080–0.397] | 6.5 |
| 18 | Z-GBAP-Des(Q1N2) | Z-(SPNIFGQWM) | 0.661[0.305–1.43] | 1.7 |
中村等人报道了基于GBAP的群体感应抑制剂的开发,所有这些抑制剂均在N端含有一个羧基苄基(Z)基团(图2C)作为保护基团。作者声称Z基团对类似物的活性没有影响。为了验证这一点,我们着手测试该基团对GBAP活性的影响。在我们的实验条件下,我们发现将Z基团添加到GBAP上(Z-GBAP,17;表1)后,其效价相对于天然GBAP提高了5倍。此外,用Z基团替换环外侧链也使效价得到了适度提升(Z-GBAP-Des(Q1N2) (18),活性提高2倍;表1)。综上所述,这些结果表明,在设计具有更强活性的基于GBAP的群体感应调节剂时,应考虑添加Z基团。
基于GBAP的超强激动剂的合理设计
本研究旨在开发具有增强活性的GBAP类似物,以用于调控Fsr群体感应系统。为此,我们首先希望优化GBAP与FsrC受体之间的结合相互作用。通过对GBAP信号分子进行构效关系研究,我们发现若干侧链和手性修饰可使肽的效价提高约5倍。这些修饰包括Gln1和Asn2位点构型的翻转(分别记为q1和n2),Pro4被丙氨酸取代(P4A),以及在N端添加Z基团。我们推测,将这些修饰组合起来会产生叠加效应,从而获得效价进一步提高的GBAP类似物。因此,我们合成了一系列双突变和三突变类似物,其中组合了q1、n2和P4A修饰,并在有或无N端乙酰基的情况下进行了评估(表2)。结果表明,在此前研究中看似可耐受的N端乙酰基修饰,当与上述任一修饰组合时,其活性均低于未加乙酰基的相应GBAP类似物。此外,某些修饰彼此不兼容(例如q1与P4A),导致所得类似物的效价较GBAP降低。最重要的是,我们鉴定出两种双突变类似物(GBAP-q1n2(19)和GBAP-n2P4A(21)),其活性显著提高(相对于GBAP,效价分别提高了11倍和24倍;表2)。
表2. 基于GBAP的理性设计超强激动剂的EC50值
| 肽# | 肽名称 | 序列 | EC50 [95% CI] (nM) | 倍数变化 |
|---|---|---|---|---|
| 19 | GBAP-q1n2 | qn(SPNIFGQWM) | 0.103[0.043–0.249] | 11 |
| 20 | GBAP-q1P4A | qN(SANIFGQWM) | 1.87[1.24–2.81] | (1.6) |
| 21 | GBAP-n2P4A | Qn(SANIFGQWM) | 0.0460[0.0300–0.0691] | 25 |
| 22 | GBAP-q1n2P4A | qn(SANIFGQWM) | 17.0[10.3–28.0] | (14.8) |
| 23 | Ac-GBAP-q1 | Ac-qN(SPNIFGQWM) | 0.314[0.112–0.880] | 3.7 |
| 24 | Ac-GBAP-n2 | Ac-Qn(SPNIFGQWM) | 1.47[0.737–2.93] | (1.3) |
| 25 | Ac-GBAP-P4A | Ac-QN(SANIFGQWM) | 1.35[0.704–2.59] | (1.2) |
| 26 | Ac-GBAP-q1n2 | Ac-qn(SPNIFGQWM) | 0.504[0.220–1.16] | 2.3 |
| 27 | Ac-GBAP-q1P4A | Ac-qN(SANIFGQWM) | 3.51[1.16–10.6] | (3.1) |
| 28 | Ac-GBAP-n2P4A | Ac-Qn(SANIFGQWM) | 0.590[0.257–1.35] | 1.9 |
| 29 | Ac-GBAP-q1n2P4A | Ac-Qn(SANIFGQWM) | 42.1[29.9–59.3] | (36.6) |
| 30 | Z-GBAP-q1n2 | Z-qn(SPNIFGQWM) | 0.0623[0.0269–0.144] | 19 |
| 31 | Z-GBAP-n2P4A | Z-Qn(SANIFGQWM) | 0.169[0.096–0.295] | 6.8 |
为了进一步优化上述两种先导肽,我们将Z-基团引入两种支架中,得到Z-GBAP-q1n2(30)和Z-GBAP-n2P4A(31)(表2)。有趣的是,Z-基团的引入使GBAP-q1n2的效价适度提高了2倍,而相同修饰在GBAP-n2P4A中却导致效价下降了3.5倍(表2)。这些结果与上述q1和P4A修饰之间的不相容性一致,表明P4A修饰无法耐受N端残基的手性反转或N端封端。图3总结了理性设计的激动剂相对于GBAP的效价变化倍数(增加或减少)。
粪肠球菌fsr群体感应回路强效激活剂和抑制剂的合理设计
基于GBAP的拮抗剂的开发
除了开发更优的激动剂外,我们还致力于开发拮抗剂,以期最终为减弱粪肠球菌感染提供一种替代治疗方法。由于已有文献报道过一种强效拮抗剂(32),我们将其结构作为支架起点,用于开发增强型抑制剂。
令人意外的是,先前报道的先导抑制剂(32)在我们的实验中几乎无活性,即使在低浓度竞争物(天然GBAP,5 nM)条件下,其IC50> 10也达到1,000 nM。该系列中所有其他尾部修饰的类似物同样无活性(表3;有关此系列化合物活性显著偏低的合成优化及验证步骤的讨论见下文)。
表3. 尾部修饰拮抗剂和基于GBAP合理设计的增强型拮抗剂的IC50值
| 肽# | 肽名称 | 序列 | IC50 [95% CI] (nM) |
|---|---|---|---|
| 32 | Z-GBAP-N5[YBzl] Q9AM11A | Z-QN(SP-YBzl-IFGAWA) | >10,000, >10,000 |
| 33 | Ac-GBAP-N5[YBzl] Q9AM11A | Ac-QN(SP-YBzl-IFGAWA) | >10,000 |
| 34 | Ac-GBAP-Des(Q1N2)N5[YBzl] Q9AM11A | Ac-(SP-YBzl-IFGAWA) | >10,000 |
| 35 | GBAP-N5[YBzl] M11A | QN(SP-YBzl-IFGQWA) | 139[61.8–313] |
| 36 | Z-GBAP-N5[YBzl] M11A | Z-QN(SP-YBzl-IFGQWA) | 227[172–299], 38.7[26.8–55.9] |
| 37 | GBAP-Q1[焦谷氨酸] N5[YBzl] M11A | [PyGlu]-N(SP-YBzl-IFGQWA) | 295[232–375] |
| 38 | Ac-GBAP-N5[YBzl] M11A | Ac-QN(SP-YBzl-IFGQWA) | 444[25–918] |
| 39 | Ac-GBAP-Des(Q1N2)N5[YBzl] M11A | Ac-(SP-YBzl-IFGQWA) | 872[520–1463] |
| 40 | Z-GBAP-q1n2N5[YBzl] M11A | Z-qn(SP-YBzl-IFGQWA) | 388[183–826] |
| 41 | Z-GBAP-n2P4AN5[YBzl] M11A | Z-Qn(SA-YBzl-IFGQWA) | >10,000 |
| 42 | GBAP-q1n2N5[YBzl] M11A | qn(SP-YBzl-IFGQWA) | 967[729–1281] |
| 43 | GBAP-n2P4AN5[YBzl] M11A | Qn(SA-YBzl-IFGQWA) | >10,000 |
| 44 | GBAP-N5[YBzl] | QN(SP-YBzl-IFGQWM) | 438[225–853] |
| 45 | Z-GBAP-N5[YBzl] | Z-QN(SP-YBzl-IFGQWM) | >1,000 |
| 46 | GBAP-q1n2N5[YBzl] | qn(SP-YBzl-IFGQWM) | >10,000 |
| 47 | GBAP-n2P4AN5[YBzl] | Qn(SA-YBzl-IFGQWM) | >1,000 |
由于我们的合成方法产率更高,并通过避免溶液中环化步骤实现了更快的合成,因此我们开发了一系列在序列中重新引入Gln9作为树脂连接点的拮抗剂类似物。单个回突变类似物(36)耐受性良好,在50 nM GBAP存在下产生的拮抗剂IC50为227 nM。为了更好地理解此前报道的抑制剂相关的构效关系,我们还开展了研究,考察了在原始支架上进行的相同尾部修饰的重要性(表3)。我们发现,在N端引入Z基团对抑制活性略有不利影响,因为去除Z基团(类似物35)导致效价略有提高,其IC50值相较于含Z基团的类似物(36)大约降低了1.5至2倍。同样地,替换不同的封端基团(乙酰基,38)使效价相对于35降低了3倍。如前所述,具有游离N端的类似物可降解形成焦谷氨酸残基。因此,也测试了该降解产物(37),结果得到的拮抗剂效价比35低约2倍。同样,去除尾部并封端肽(39)导致该系列中活性最低的类似物(与35相比效价损失约6倍)。综合来看,这些结果解释了为何所有基于已发表的骨架(32-34)所准备的肽类似物均被发现无活性。此处观察到的趋势表明,只有游离N端的类似物(我们未能合成,见下文)才会比母体肽支架(32)更具活性。
在增强激动剂研究过程中,我们发现了几种可显著提高GBAP与FsrC受体结合亲和力的修饰。我们假设,在我们的树脂相容性抑制剂(35和36)中引入相同的修饰将增强这些抑制剂的活性。然而,不幸的是,我们发现事实并非如此。基于我们两种最有效激动剂所构建的四种拮抗剂类似物均不如其母体化合物(35或36;表3)活跃。引入q1n2修饰并带有Z基团的拮抗剂类似物(40)比36的效价低2倍,而引入n2和P4A修饰并带有Z基团的拮抗剂类似物(41)则完全丧失了抑制活性。类似地,引入q1n2修饰但不含Z基团的类似物(42)比35的效价约低7倍,而引入n2P4A修饰但不含Z基团的类似物(43)也像41一样完全丧失了抑制活性。尽管基于上述观察到的P4A与Z基团修饰之间的不相容性(表2),41活性降低是可以预见的,但40与36相比活性降低,以及42和43与35相比活性降低的现象表明,尾部修饰与将GBAP转化为竞争性抑制剂所需的环状修饰并不兼容。
产生拮抗剂所需的最小修饰
在我们发现将先前报道的抑制剂(32)重新引入Gln9得到化合物36后,其效价提高了>50倍,我们随即着手确定将GBAP转化为抑制剂所需的最小修饰。以36为起点,该类似物是与先前报道的抑制剂(32)最接近且适用于我们完全在树脂上合成路线的化合物,我们首先将Met11回复突变为45。该类似物保留了其抑制特性,但效价比36低>4倍。由于从36中去除Z基团得到35后,效价略有提高且仍保持抑制活性,因此我们推测从45中去除Z基团将获得更优的抑制剂。事实上,44的效价比45高出>2倍(表3)。有趣的是,该类似物与GBAP仅在一个残基上存在差异(Asn5至苄基酪氨酸(YBzl;图2D)),结果发现其为部分激动剂,因其未能完全消除群体感应活性,而是将QS活性降低了约80%。这一结果强调了该修饰在将GBAP转化为抑制剂过程中的重要性,同时也表明需要第二个修饰才能彻底消除激动活性。此外,拮抗剂文库的综合结果表明,尾部区域对拮抗活性的重要性高于对激动活性的影响,因此对该区域的修饰耐受性较低(比较表1和表2与表3的活性趋势)。
由于我们发现苄基酪氨酸修饰足以将GBAP转化为部分抑制剂,而要将肽转化为完全竞争性抑制剂则需要额外的修饰,因此我们尝试将增强的激动剂修饰(q1n2或n2P4A)引入该最小修饰的支架中,分别得到46和47。遗憾的是,这两种类似物的效力均低于其母体支架(44),进一步强调了尾部修饰与苄基酪氨酸环修饰之间的不相容性。
合成难以制备的基于GBAP的类似物
我们立即发现,我们完全采用的树脂上合成方法与32及其类似物所基于的支架不兼容,因为我们的方法所使用的两个树脂连接点(天冬酰胺5和谷氨酰胺9)在设计中已被移除(方案S-1)。因此,需要一种替代的合成方法。最初,我们遵循了文献报道的拮抗剂合成中引用的方法(方案S-1)。然而,在我们的实验条件下,该方法产率极低,并且在肽链多个位点发生了严重的差向异构化。此外,如上所述,化合物32的抑制活性远低于预期。
我们推测,合成方法(特别是困难的环化步骤)可能是导致活性不一致的原因。为了帮助解决产率问题以及观察到的效价不一致的情况,我们开发了一种替代的合成方法,该方法引入了我们完全在树脂上合成策略中的一些方法,即先在树脂上进行酯形成,然后通过酰胺形成在溶液中进行简便的环化(方案S-1)。使用这种替代方法制备的肽(32)仍然无活性,表明合成方法的差异并不是我们在实验中可重复观察到的显著低活性的原因。此外,当与我们的完全在树脂上合成相容的最相似肽(36)使用与制备32相同的方法重新合成时,36仍保持其效价。综合来看,这些结果进一步验证了我们所采用的合成方法的可靠性,以及我们在实验中观察到32缺乏活性的真实性。
尽管我们引入的合成修饰使我们能够制备大多数计划中的肽,包括32,但由于溶液中环化后脱除N端保护基所涉及的额外步骤导致产率极低,我们仍无法制备一种缺少N端封端基团的计划类似物(参见SI中对此特定肽所用方法的描述)。此外,虽然改进的合成方法已将异构体的生成减少到更合理的水平,但仍存在至少一种显著且难以去除的污染物(一种异构体)(该异构体经过测试,发现无活性)。
在表型检测中评估先导拮抗剂
尽管我们的报告基因检测已证实这些肽能够调节群体感应反应,但我们还想验证这种调节是否可转化为对治疗上重要表型的影响。为此,我们测试了先导拮抗剂35在野生型粪肠球菌细胞(菌株TX4002)中减弱生物膜形成的能力。由于生物膜形成也可能受到其他通路的影响,我们纳入了一个群体感应失活菌株(TX5266)作为对照(图4和S-1)。我们发现,向细胞中添加外源性GBAP可使生物膜形成量增加约30-50%。然而,无论是否加入外源性GBAP,只要加入浓度比其IC50高5倍的化合物35,就足以将生物膜形成减少至群体感应失活菌株的水平(减少约30-50%)。这些结果表明,即使存在足量的天然激动剂以充分诱导群体感应反应,该拮抗剂仍能完全消除依赖于群体感应的生物膜形成。
结论
我们利用当前及以往关于GBAP信号的构效关系知识,合理设计出具有增强活性的基于GBAP的群体感应调节剂。为此,我们开发、优化并比较了三种用于构建GBAP类似物的合成路线。结果表明,完全采用树脂上合成方法在效率、纯化难易程度和总收率方面均优于两种基于溶液的环化方法。
我们设计的增强型激动剂库获得了多个活性显著提升的类似物(效价提高>20倍)。该库还揭示了若干彼此不兼容的修饰,这些修饰组合后会导致效价下降。推测这种不兼容性可能是由于各修饰引起的构象变化引入了空间位阻所致,但还需进一步的结构研究加以验证。图5总结了从激动剂库中获得的关键构效关系结论。
关于基于GBAP的竞争性抑制剂,我们发现单个修饰(N5[YBzl])足以将GBAP转化为一种在纳摩尔范围内具有IC50值的强效抑制剂,即使在存在显著浓度天然竞争物的情况下也是如此。该结果强调了验证所有修饰的必要性,以正确归因所观察到的构效关系(SARs)与其相关修饰(先前使用反向丙氨酸扫描方法的研究能够识别出这种意外的修饰具有重要性,但更强调丙氨酸取代对所观察到的SARs的贡献)。我们对基于GBAP的拮抗剂进行的系统分析揭示了具有更强活性的抑制剂,其活性相比先前报道的先导化合物(32)提高了>50倍。因此,这些新类似物是设计具有潜在治疗意义的基于GBAP的群体感应抑制剂的优势骨架。事实上,我们的先导抑制剂35能够在野生型粪肠球菌菌株中完全抑制致病表型——生物膜形成,使其水平达到与群体感应失活菌株相当的程度。
综合本研究获得的构效关系见解,提示了几个可进一步改善结合能力的区域,或可进行耐受性良好的修饰,这些修饰可能有助于提高肽类似物的稳定性。首先,在耐受性位点(如Phe7)引入额外的D-氨基酸,可提高肽抵抗蛋白酶降解的稳定性。稳定性的提升可能抵消其伴随的轻度效价损失,尤其是在本身已具有高活性的肽中。在Phe7位点引入D-色氨酸取代,其活性接近天然肽,可能在几乎不影响效价的前提下显著提高肽的稳定性。其次,环外尾部区域具有显著改变激动剂和拮抗剂活性的能力。这可通过改变环外侧链残基的立体化学或通过N端封端来实现。此类旨在进一步提高激动剂和拮抗剂活性的实验,以及深入的结构研究以阐明驱动GBAP-FsrC结合并导致FsrC激活的分子机制,目前正在本实验室进行,将在适当时候予以报道。

















 3922
3922

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









