







在当今的科学研究中,AI for Science 迅速成为解决复杂问题的重要工具。然而,如何将科学原理融入机器学习建模,以在有限训练数据下提升模型的预测性能,仍是一个重大挑战。在此背景下,深势科技推出了名为 Uni-pKa 的创新框架,将热力学原理融入机器学习建模中,并结合预训练-微调的训练策略,充分利用了不同质量的数据,实现了对酸解离常数(pKa)的高精度预测。

有机小分子的酸碱性对其在生物体内展现的药效、代谢和毒性有深远的影响[1]。精确的药物活性预测(如自由能微扰计算)必须定量地考虑分子在水溶液中的酸碱平衡[2]。因此,预测酸性电离平衡常数(pKa)是计算机辅助药物设计中的重要任务。随着 AI for Science 的发展,数据驱动的 pKa 预测方法以其高通量和可接受的精度,从基于经验或传统计算化学的方法中脱颖而出[3]。然而,在全原子模拟的结构准备、构效关系分析、自由能计算修正等备受关注的需求中,由于要同时处理分子中多个化学基团互相耦合的酸碱平衡,更精确的 pKa 预测方法面临巨大的挑战。这些方法由于过度简化了化学平衡网络并忽略了热力学约束,预测结果常常自相矛盾[4]。因此,领域亟需一个快速、精确且自洽的工具来预测复杂类药分子的整体酸碱性质。
2024年7月,深势科技在 JACS Au 上发表封面文章 Bridging Machine Learning and Thermodynamics for Accurate pKa Prediction[5],介绍了基于热力学原理和预训练分子大模型的高精度 pKa 预测方法——Uni-pKa(可点击文末“阅读原文”查看全文)。文章提出了从数据采集到模型设计一以贯之的质子化系综(protonation ensemble)框架:配备了质子化状态枚举工具,深入挖掘公开 pKa 数据集的化学内涵;从自由能预测的第一性视角处理 pKa 问题,使全面且自洽的酸碱平衡建模成为可能。基于预训练的三维分子大模型 Uni-Mol[6],多种预训练任务和微调的配合使 Uni-pKa 在多个类药分子数据集上取得了最佳精度。这项工作得益于先进的机器学习模型与严格物理化学原理的有机结合,适用于复杂分子酸碱平衡建模的多种任务,提高了数据驱动的 pKa 预测方法的准确性与可靠性。


建模方法
质子化系综是指一个分子在水溶液中通过酸式电离(去质子化)或碱式电离(质子化)能够得到的所有化学结构。Uni-pKa 源自以下热力学原理:无论分子中的酸碱平衡怎样复杂,其所有信息可被质子化系综中各状态的自由能完全确定。在这一视角下,单个位点、单个电离反应的 pKa 对应于质子化系综中两分子间的自由能差,而一些实验方法测得的 pKa 对应于质子化系综中两组分子之间的粗粒化自由能差,各电离形态的分布则是质子化系综在给定 pH 下的 Boltzmann 分布[7]。这三条论断分别服务于三个与 pKa 相关的主要任务:micro-pKa 预测、macro-pKa 预测、质子化状态分布预测。因此,只要能找到一条联结 pKa 数据与质子化系综的自由能阶梯之间的纽带,就能一举实现复杂分子全面而自洽的酸碱平衡建模。为此,Uni-pKa 完成了三项工作:
1. 解译 pKa 数据,对应到质子化系综中的分子
2. 设计一个分子机器学习模型,目标为预测质子化系综中分子的自由能
3. 根据热力学理论把 pKa 和自由能定量地联系起来,使模型能够在 pKa 数据上学习自由能
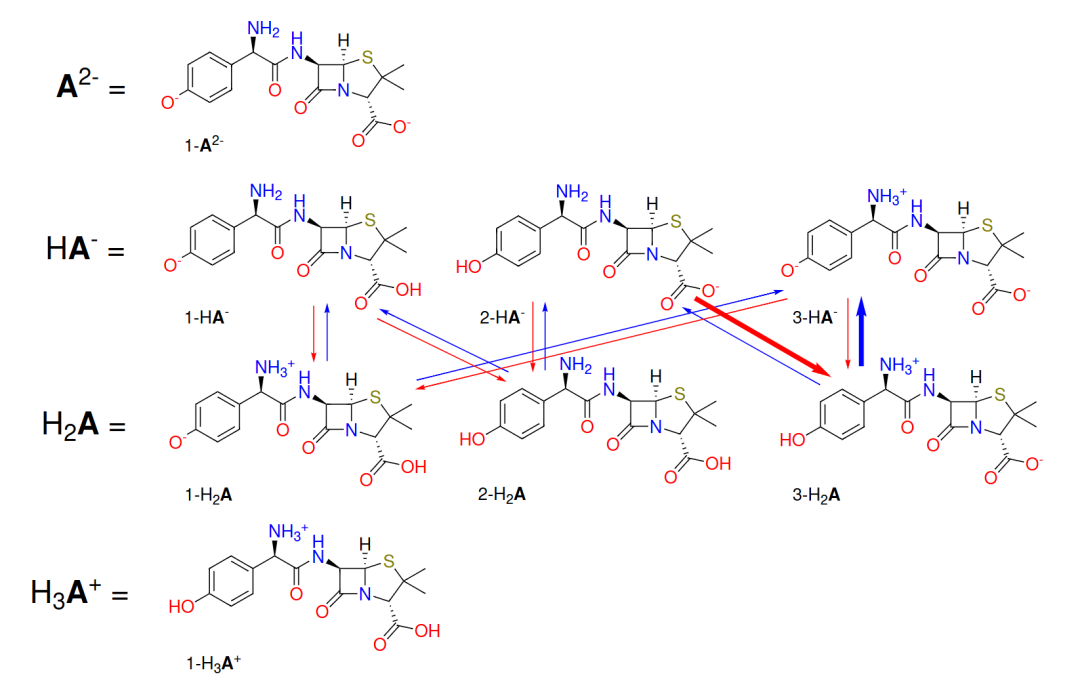
药物分子阿莫西林的质子化系综一瞥

训练与推理策略
基于以上思路,Uni-pKa 的工作流分为以下几部分:
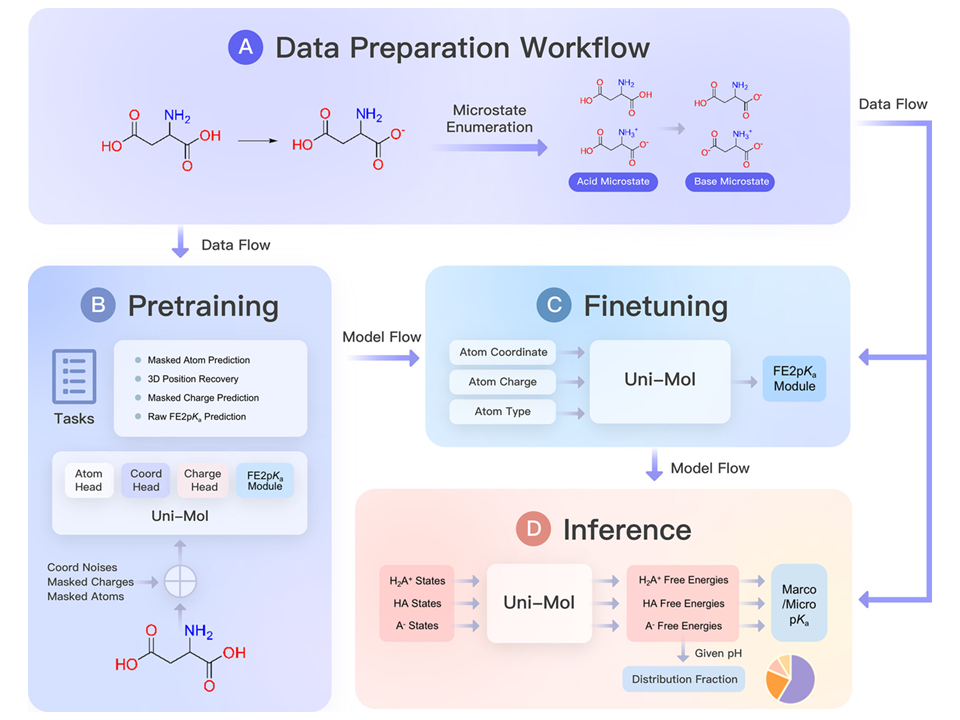
基于质子化系综理论的 Uni-pKa 框架
1. 我们设计了质子化状态枚举工具,通过识别和匹配化学模板中的可离子化位点,实现从单一分子到完整质子化系综的转换。借助这一工具,我们基于数据集的引用来源将不同实验方法测得的 pKa 解读为 micro-pKa 或 macro-pKa,并转译为质子化系综中参与电离反应的具体分子结构。
2. 我们采用了 Uni-Mol 这一强大、通用的分子机器学习模型作为自由能预测器,并依据化学经验定制了原子电荷描述符。然而精确的 pKa 实验数据非常有限;尽管质子化状态枚举扩充了数据集中的信息量,但为了保证模型的泛化能力,仅凭现有实验数据上的直接训练覆盖的化学空间是远远不够的。于是,Uni-pKa 采用了预训练-微调这一范式——首先基于计算软件预测的 pKa 数据让模型学习从分子结构到 pKa 数值的基本映射关系,然后再使用实验测量的 pKa 数值来驱动模型形成有效的 pKa 预测能力,具体的:
a. 预训练在超过110万个数据点、由软件预测的 ChEMBL 低精度 pKa 数据集[8]上进行。预训练策略结合了预测低精度 pKa 值的弱监督任务和 3D 位置恢复、掩码原子预测和掩码电荷预测三个自监督任务。
b. 模型微调在有8200多个数据点,整理自实验文献的 Dwar-iBonD 高精度 pKa 数据集上进行,这些数据来自 DataWarrior 软件示例数据[9]和 iBonD 键能数据库[10],涵盖了从简单羧酸到复杂生物碱和卟啉衍生物的广泛化学空间。微调进一步提高了模型的预测精度。同时为了确保与预训练阶段的一致性,我们在微调过程中使用了相同的数据准备工作流程和 pKa 预测任务设置。
3. 我们从热力学理论出发,利用自由能与化学平衡之间的定量关系设计了 FreeEnergy2pKa 的输出模块和损失函数。这一模块应用于预训练阶段的弱监督任务和整个微调流程,使得模型从枚举好的质子化系综中的分子结构出发端到端地学习 pKa 数据,并将模型的输出自然地对应到分子的自由能。
训练完毕后,Uni-pKa 将输入分子枚举为质子化系综,由 Uni-Mol 内核预测自由能,最后经过 FreeEnergy2pKa 的处理,将宏观 pKa 预测、微观 pKa 预测和分布分数预测统一在了一套推理框架下。
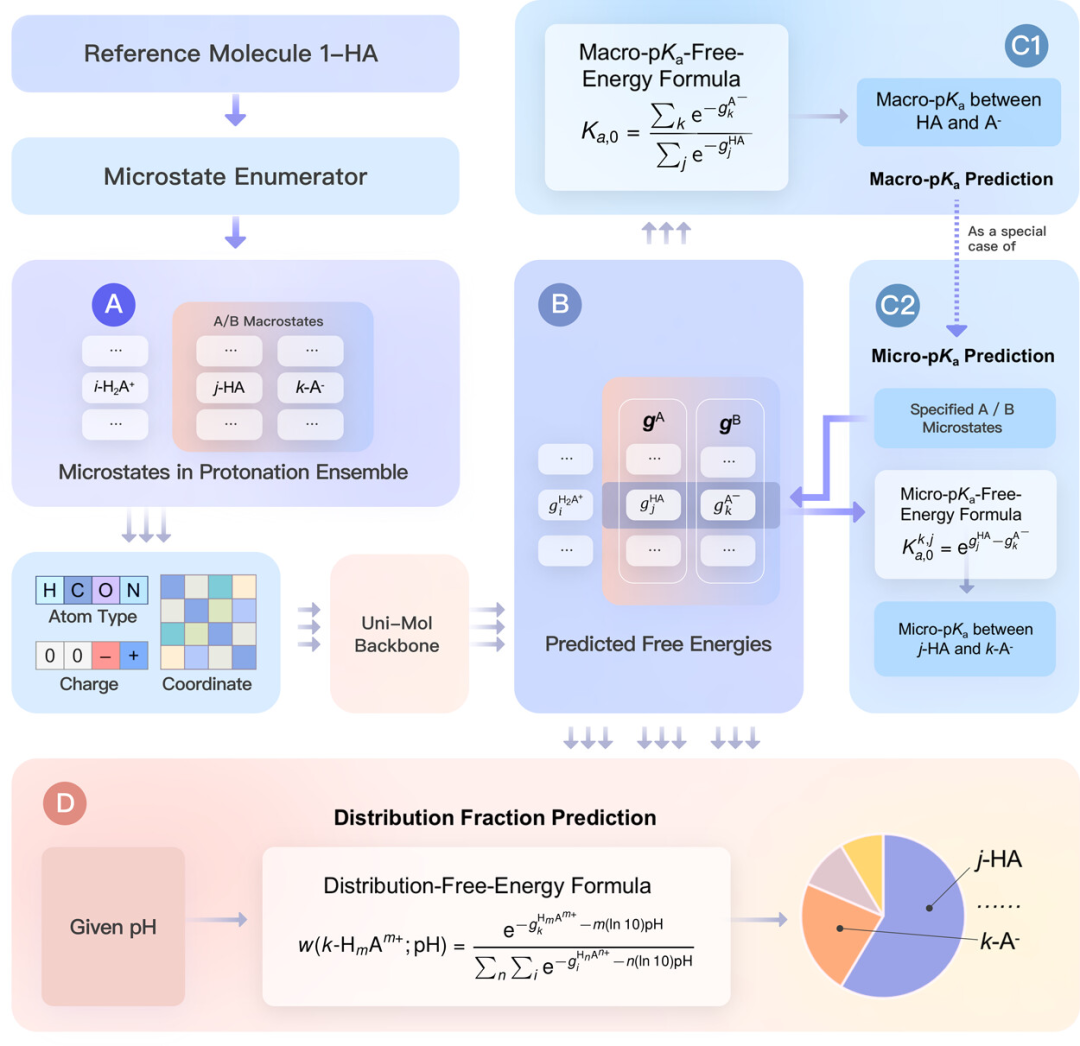
以 Uni-Mol 为核心的自由能预测模型适用于多种 pKa 预测任务

预测精度
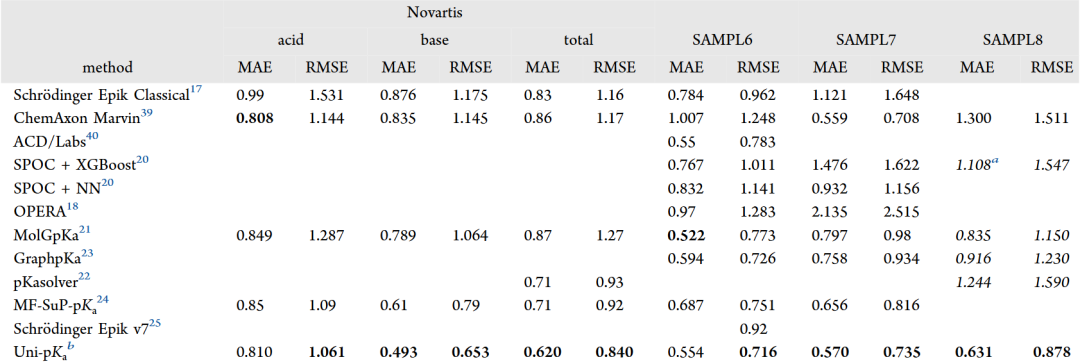
Uni-pKa 在多个类药分子测试集上达到了同类方法中的最佳精度
在最新的 SAMLP8 pKa 预测挑战数据集[11]中,Uni-pKa 达到了 MAE=0.631,RMSE=0.878 的精度,与此前的公开 pKa 推理工具相比具有至少0.2个 pKa 单位的优势。
在其他几个常用的基准数据集上,Uni-pKa 的表现也优于现有的化学信息学方法,显著提高了预测精度。具体来说,在 Novartis 数据集[12]上,Uni-pKa 的平均绝对误差(MAE)为0.620,表现显著优于其他方法,如 Schrödinger Epik Classical[13] 的0.83和 ChemAxon Marvin[14] 的0.86。在 SAMPL6[15] 和 SAMPL7[16] 数据集上,Uni-pKa 也同样优于其他方法。
在预测类药分子复杂的化学修饰对 pKa 的影响时,Uni-pKa 同样表现出色。在 Schrödinger 的量子化学方法 Jaguar 发表的示例数据[17]上,Uni-pKa 的精度能与简单流程的量子化学方法媲美,且在 Novartis 数据集上每秒平均预测28个宏观 pKa 的速度与耗时冗长的量子化学计算相比有显著优势。

热力学自洽性
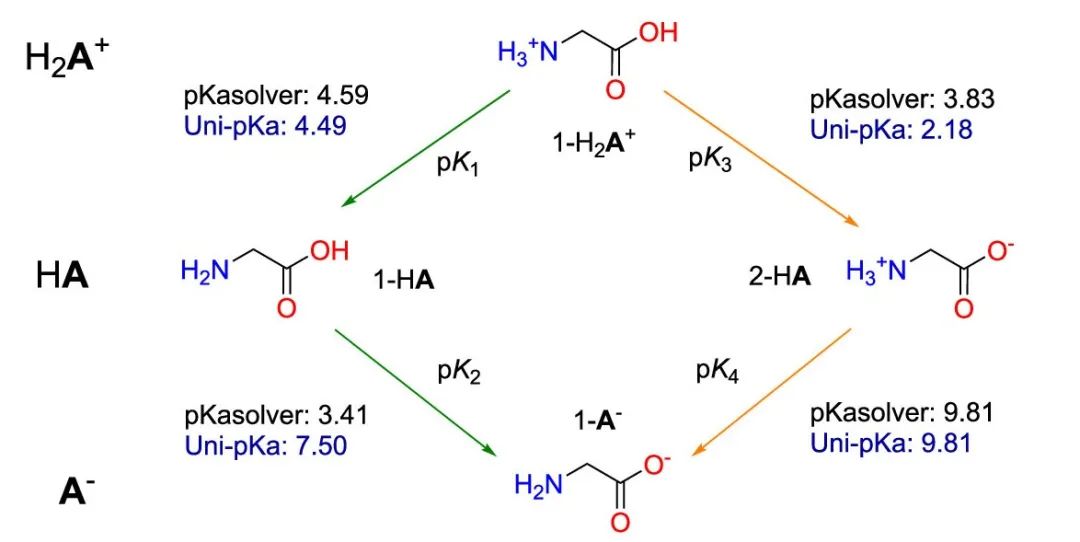
Uni-pKa 在经典生物化学现象:谷氨酸的两性离子问题上展现出的热力学自洽性,沿绿色路线和黄色路线的 pKa 之和相等
Uni-pKa 善于处理多个电离位点和连续电离的复杂情况。我们用 Uni-pKa 和较为先进的深度学习方法 pKasolver[18] 研究了甘氨酸的经典的电荷分离现象:如图所示,电中性形态的甘氨酸有分子形态和电荷分离的内盐形态,实验表明后者是甘氨酸在等电点附近的主要形态,这一现象造就了氨基酸乃至多肽和蛋白质的极性,是众多的生物化学结构和功能的基础。为了定量描述内盐形态和电中性形态的比例,可以借助正电形态与两种电中性形态之间的 pKa 之差,也可以用两种电中性形态与负电形态之间的 pKa 之差——两种描述方式来自酸式电离和碱式电离的两个视角,理应给出完全一致的结果,即图中 pK1 - pK3 = pK4 - pK2。我们发现 pKasolver 的预测结果不满足这一条件,而 Uni-pKa 给出了殊途同归的自洽结果,从而能够唯一确定给定 pH 条件下甘氨酸各形态的比例,这一自洽性是 Uni-pKa 自由能建模背后的热力学约束所保证的。因此,Uni-pKa 中融入的物理化学原理使得它在处理复杂分子的应用场景时更加严谨、可靠。

总结
Uni-pKa 为经典的 pKa 预测问题提供了一个交叉学科的解决方案:它基于物理化学原理进行科学问题的深入剖析,并利用机器学习大模型高效处理科学数据,建立了一个准确且可靠的复杂化学平衡建模框架。在 Uni-Mol 基座模型强大的分子表示学习能力支持下,Uni-pKa 通过多种预训练任务和微调策略,实现了在多个类药分子数据集上的最佳预测精度。这一方法不仅提高了 pKa 预测的准确性和可靠性,还展示了将热力学原理与机器学习技术相结合的潜力。
Uni-pKa 是 AI for Science 的成功实践,不仅为 pKa 预测领域带来了新的突破,还为将物理知识与机器学习结合应用于更广泛的科学研究提供了宝贵的经验。这一框架展示了如何充分应用不同质量的数据,来更好地解决科学数据稀少的问题。

开源仓库、数据集和APP
Uni-pKa 现已开源!在遵从 Apache-2.0 协议的前提下,用户可从深势科技 GitHub 仓库获得 Uni-pKa 的质子化状态枚举器和 Uni-Mol 模型训练、推理的源代码,将 Uni-pKa 框架应用于自己的研究和工作中。
Uni-pKa 官方仓库:
https://github.com/dptech-corp/Uni-pKa
如果您想使用 Uni-pKa 工作中基于质子化系综理论整理和扩充数据集,可以在科学智能广场(AIS Square)下载。其中除了 pKa 数据之外,还包含 ChEMBL 数据集中超过三百万个分子在 xTB-GFN2 水平下的几何优化结构和原子电荷数据。
Uni-pKa 数据集:
https://www.aissquare.com/datasets/detail?pageType=datasets&name=Uni-pKa-Dataset
如果您想使用训练好的 Uni-pKa 模型预测分子在给定 pH 值下的质子化状态及其分布,您可以在深势科技Bohrium® App上提交您感兴趣的分子,平台会将您的任务自动分配计算资源,推理并返回结果。
Uni-pKa App:
https://bohrium.dp.tech/apps/uni-pka
参考文献
[1]: Manallack, D. T.; Prankerd, R. J.; Yuriev, E.; Oprea, T. I.; Chalmers, D. K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42 (2), 485– 496.
[2]: De Oliveira, C.; Yu, H. S.; Chen, W.; Abel, R.; Wang, L. Rigorous free energy perturbation approach to estimating relative binding affinities between ligands with multiple protonation and tautomeric states. J. Chem. Theory Comput. 2018, 15 (1), 424– 435.
[3]: Jialu, W.; Kang, Y.; Pan, P.; Hou, T. Machine learning methods for pK(a) prediction of small molecules: Advances and challenges. Drug Discovery Today 2022, 27, 103372.
[4]: Johnston, R. C.; Yao, K.; Kaplan, Z.; Chelliah, M.; Leswing, K.; Seekins, S.; Watts, S.; Calkins, D.; Elk, J. C.; Jerome, S. V.; Repasky, M. P.; Shelley, J. C. Epik: pK(a) and protonation state prediction through machine learning. J. Chem. Theory Comput. 2023, 19 (8), 2380– 2388.
[5]: Luo, W.; Zhou, G.; Zhu, Z.; Yuan, Y.; Ke, G.; Wei, Z.; Gao. Z.; Zheng, H. Bridging Machine Learning and Thermodynamics for Accurate pKa Prediction. JACS Au. 2024, DOI: 10.1021/jacsau.4c00271
[6]: Zhou, G.; Gao, Z.; Ding, Q.; Zheng, H.; Xu, H.; Wei, Z.; Zhang, L.; Ke, G. Uni-Mol: A universal 3D molecular representation learning framework. In Paper presented at the Eleventh International Conference on Learning Representations , 2023, https://openreview.net/forum?id=6K2RM6wVqKu.
[7]: Gunner, M. R.; Murakami, T.; Rustenburg, A. S.; Işık, M.; Chodera, J. D. Standard state free energies, not pK(a)s, are ideal for describing small molecule protonation and tautomeric states. J. Comput.-Aided Mol. Des. 2020, 34, 561– 573.
[8]: Gaulton, A.; Bellis, L. J.; Patricia Bento, A.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40 (D1), D1100– D1107
[9]: Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55 (2), 460– 473
[10]: Internet Bond-energy Databank (pKa and BDE)–iBonD Home Page , 2017. https://ibond.las.ac.cn/
[11]: Nandkeolyar, A.; Bahr, M. N.; Mobley, D. L. Insights from the SAMPL8 physical properties blind prediction challenge. Biophys. J. 2023, 122 (3), 423a
[12]: Liao, C.; Nicklaus, M. C. Comparison of nine programs predicting pK(a) values of pharmaceutical substances. J. Chem. Inf. Model. 2009, 49 (12), 2801– 2812
[13]: Shelley, J. C.; Cholleti, A.; Frye, L. L.; Greenwood, J. R.; Timlin, M. R.; Uchimaya, M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681– 691
[14]: Prediction of dissociation constant using microconstants. https://docs.chemaxon.com/display/docs/attachments/attachments_1814016_1_Prediction_of_dissociation_constant_using_microconstants.pdf
[15]: Işık, M.; Rustenburg, A. S.; Rizzi, A.; Gunner, M. R.; Mobley, D. L.; Chodera, J. D. Overview of the SAMPL6 pK(a) challenge: evaluating small molecule microscopic and macroscopic pK(a) predictions. J. Comput.-Aided Mol. Des. 2021, 35 (2), 131– 166
[16]: Bergazin, T. D.; Tielker, N.; Zhang, Y.; Mao, J.; Gunner, M. R.; Francisco, K.; Ballatore, C.; Kast, S. M.; Mobley, D. L. Evaluation of log P, pK(a), and log D predictions from the SAMPL7 blind challenge. J. Comput.-Aided Mol. Des. 2021, 35 (7), 771– 802
[17]: Bochevarov, A. D.; Watson, M. A.; Greenwood, J. R.; Philipp, D. M. Multiconformation, density functional theory-based pK(a) prediction in application to large, flexible organic molecules with diverse functional groups. J. Chem. Theory Comput. 2016, 12 (12), 6001– 6019
[18]: Mayr, F.; Wieder, M.; Wieder, O.; Langer, T. Improving small molecule pK(a) prediction using transfer learning with graph neural networks.. Front. Chem. 2022, 10, 866585
上下滑动查看更多
关于深势科技
深势科技是“AI for Science”科学研究范式的引领者和践行者。AI for Science 即运用 AI 学习一系列的科学原理和科学知识,并进一步解决科学研究和工业研发领域的关键问题。
深势科技依托在交叉学科领域的深耕,构建了“深势 · 宇知®”AI for Science 大模型体系,将众多学科的科研方法从“实验试错 / 计算机”时代带入了“预训练模型时代”,并以“微尺度工业设计与仿真”为切入点,打造了 Bohrium®玻尔®科研空间站、Hermite® 药物计算设计平台、RiDYMO® 难成药靶标研发平台及 Piloteye® 电池设计自动化平台等科研和工业研发基础设施,形成了 AI for Science 的“创新 - 落地”链路和开放生态,赋能“千行百业”,为人类经济发展最基础的生物医药、能源、材料和信息科学与工程研究打造新一代工业设计与仿真系统。
深势科技是国家高新技术企业、国家专精特新“小巨人”企业,在北京、上海、深圳等城市布局研发中心。科研技术团队由中国科学院院士领衔,汇集了超百位数学、 物理、化学、生物、材料、计算机等多个领域的优秀青年科学家和工程师,其中博士及博士后在公司成员中占比超过 35%。核心成员获得过2020年全球高性能计算领域的最高奖项“戈登贝尔奖”,相关工作入选2020年中国十大科技进展和全球 AI 领域十大技术突破。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









