







Lin, X., Tian, T., Wei, Z. et al. Clustering of single-cell multi-omics data with a multimodal deep learning method. Nat Commun 13, 7705 (2022).
论文地址:https://doi.org/10.1038/s41467-022-35031-9
代码地址:https://github.com/xianglin226/scMDC/releases/tag/v1.0.0
目录
摘要
单细胞多模态测序技术旨在同时分析同一细胞中的不同模态数据。这为在单细胞水平联合分析多模态数据以识别不同的细胞类型提供了独特的机会。正确的聚类结果对于后续复杂的生物学功能研究至关重要。然而,将不同的数据源结合起来进行单细胞多模态数据的聚类分析仍然是一个统计和计算方面的挑战。在此,我们开发了一种新颖的多模态深度学习方法,scMDC,用于单细胞多组学数据的聚类分析。scMDC 是一种端到端的深度模型,能够显式表征不同的数据源,并联合学习深度嵌入的潜在特征以进行聚类分析。广泛的模拟和真实数据实验表明,scMDC 在不同的单细胞多模态数据集上优于现有的单细胞单模态和多模态聚类方法。其线性扩展的运行时间使 scMDC 成为分析大型多模态数据集的有前景的方法。
引言
单细胞RNA测序(scRNA-seq)可以为单个细胞内部的状态提供高分辨率的图谱。基于scRNA-seq技术,近年来开发了许多多模态测序技术,能够在单细胞中联合分析多种模态数据。例如,CITE-seq(转录组和表面蛋白联合测序) 和 REAP-seq(RNA表达与蛋白测序) 技术已经实现了对mRNA表达和细胞表面蛋白的同时定量。这些技术利用现有的单细胞测序平台,如10X Genomics Chromium系统,通过抗体衍生标签(ADT)计数来量化细胞表面蛋白的丰度。ADT标记和DNA条形码微珠与单细胞共同包裹在液滴中进行测序。此外,REAP-seq结合了DNA条形码抗体和现有的scRNA-seq方法,从而测量基因和细胞表面蛋白的表达水平。
除了研究单细胞的转录组和表面蛋白,最近发展起来的单细胞可转座酶可接触染色质测序(scATAC-seq)技术使我们能够测量单细胞的染色质可及性。该技术通过使用超活性Tn5转座酶同时标记和片段化开放染色质区域的DNA序列,从而识别基因组中的开放染色质区域。通过分析染色质可及性特征(如调控基因表达的转录因子),scATAC-seq可以探究特定细胞类型的生物学活动。进一步地,一些单细胞多组学技术(如SNARE-seq和10X Single-Cell Multiome ATAC + Gene Expression,简称SMAGE-seq)能够在单细胞中联合分析染色质可及性和基因表达数据。
总体而言,这些多模态测序技术为单细胞的复杂和全面表征提供了可能。然而,这也对多模态数据的整合和下游分析(如聚类分析)提出了更高的需求。在多模态数据中,不同模态提供的生物信息通常是互补的,并且各自具有优势与局限性。例如,CITE-seq技术的ADT模态专注于表面蛋白,并显示出较低的缺失率(最高可达12%),能够可靠地量化细胞活动。相比之下,其对应的mRNA数据中超过80%甚至90%的条目为零。尽管如此,mRNA的全转录组信息可以捕获更全面的细胞类型信息,尽管其高缺失率和稀疏信号会对聚类分析构成挑战。
类似地,scATAC-seq数据通过提供染色质可及性信息,与mRNA数据形成互补关系。通过整合多模态信息,我们可以获得更高分辨率的细胞分类结果。
单细胞聚类分析的现状
聚类分析是单细胞研究中的一个关键步骤。研究人员通过聚类结果,可以在细胞类型或亚型水平上探索生物学活动。许多针对scRNA-seq数据的聚类方法已被开发,例如Tscan、Seurat和SC3等。然而,这些传统方法未能充分利用多模态数据的潜力,因此难以应用于多模态数据的聚类分析。
近年来,一些针对CITE-seq数据的聚类方法开始出现,例如scDCC、BREM-SC和CiteFuse等。其中,BREM-SC通过分层贝叶斯混合模型联合建模scRNA-seq和ADT数据。然而,它的假设(如数据分布形式)限制了其适用性,同时在应对大规模数据时计算效率较低。相比之下,Seurat V4提出的加权最近邻(WNN)算法和Specter通过图构建方法进行聚类,效率更高,但仍未解决scRNA-seq数据中的缺失问题。
深度学习方法的应用
此外,一些研究集中在不同模态的联合嵌入学习,以提升下游分析(包括聚类)的性能。例如,TotalVI和Cobolt使用变分自编码器(VAE)联合建模多模态数据,并学习单细胞数据的联合表示。然而,这些方法未针对聚类进行优化。
为了解决上述问题,我们提出了一种基于深度学习的多模态单细胞聚类模型——Single Cell Multimodal Deep Clustering(scMDC)。scMDC采用多模态自动编码器框架,通过ZINB(零膨胀负二项分布)损失函数应对scRNA-seq数据的高缺失率,同时引入基于KL散度的损失函数以优化潜在特征学习。整个模型包括自动编码器、KL损失和深度K均值聚类,能够同时完成多模态数据的整合和聚类分析。实验表明,scMDC在CITE-seq和SMAGE-seq数据上的表现优于现有方法,并且在处理大规模数据时具有高效性。
总之,scMDC为多模态单细胞数据的聚类分析提供了一个高效且性能优越的工具。
模型
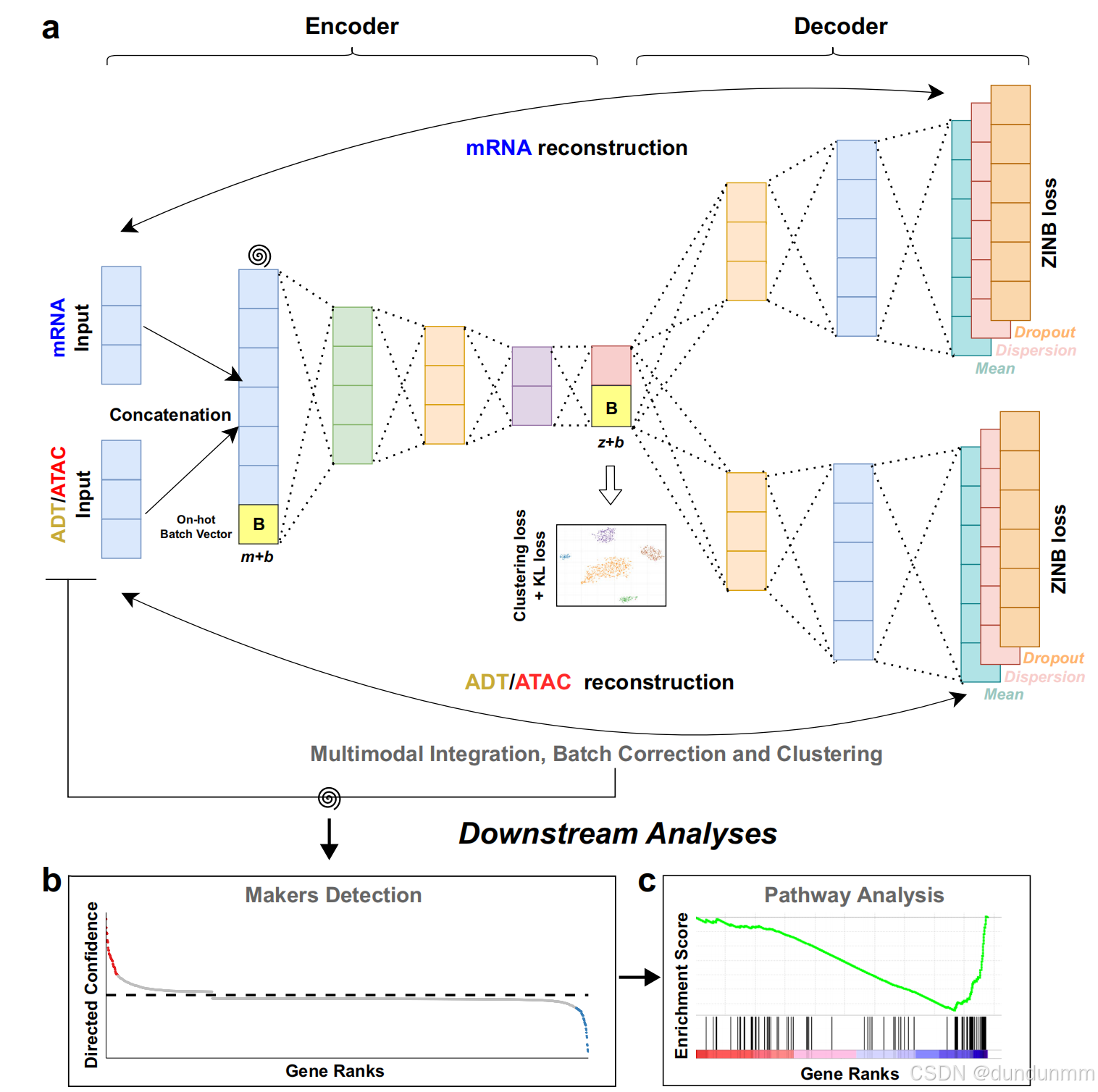
scMDC 的架构图。scMDC 为多模态数据的连接数据设置了一个编码器,并为每种模态分别配置了两个解码器 (图 a)。它可以用于聚类 CITE-seq 数据和 10x 单细胞多组学 ATAC + 基因表达 (SMAGE-seq) 数据。螺旋符号表示人工添加的数据噪声。对于多批次数据集,scMDC 将以条件自编码器的方式运行。一个独热编码的批次向量 B(维度为 b)将分别与编码器的输入特征(原始特征维度为 m)和解码器的输入特征(潜在特征维度为 z)连接。这种设计用于批次效应校正。
scMDC 学习数据的潜在表示 Z(维度为 z),实现不同模态的整合。在 Z上实现了深度 K-means 算法和 KL损失。基于聚类结果,scMDC 利用 ACE 模型来检测不同聚类中的标志物 (图 b)。随后,可以基于 ACE 所学习的基因排序进行通路分析 (图 c)。
讨论
我们提出了 scMDC ——一种用于不同单细胞多组学数据聚类分析的多模态深度学习方法。scMDC 通过多模态自编码器联合建模 mRNA 和 ADT/ATAC 数据。在自编码器的瓶颈层进行深度 K-means 聚类,同时利用 KL 损失促进不同细胞群的分离。scMDC 是一个端到端的深度模型,其所有组件可以同时优化。
目前现有的 CITE-seq 数据聚类方法要么采用浅层贝叶斯模型(如 BREM-SC),要么通过组合 mRNA 和 ADT 的两个基于距离的图(如 CiteFuse 和 Seurat)整合不同数据来源的信息。然而,这些方法并未显式建模 mRNA 和/或 ADT 计数数据中的 dropout 事件和过度离散性。我们的实测数据结果表明,基于多模态的深度学习方法能够更高效、更准确地表征 CITE-seq 和 SMAGE-seq 的不同计数数据来源。
聚类结果对于后续分析(如差异表达分析和基因集富集分析)至关重要。我们采用基于深度学习的差异表达算法,通过其被分配到目标聚类的置信度对基因进行排序。基于排序的基因列表,可以进行基因集富集分析(GSEA),以从功能层面对细胞类型进行分析。相比于传统方法(如 Wilcoxon 检验和 DESeq2),这种深度差异表达方法的优势已被 Lu 等人证明。
借助 GPU 的加速,scMDC 在分析大规模多组学数据集时效率非常高。综合所有结果,我们认为 scMDC 是分析单细胞多组学数据的一种具有前景的方法。
这一篇师单细胞多组学数据聚类的比较早的论文了,可以以这一篇为起点,下一步实现代码复现和实验结果分析。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









