






来源公众号:分子动力学
必读文献
PART-01
派言派语 写在前面
近年来随着分子动力学模拟方法在化学、生物及相关学科研究中的地位逐渐凸显,其得到越来越多的关注。
自1977年Karplus等人首次完成牛胰蛋白酶抑制剂的分子动力学模拟以来,Web of Science数据库中题目或摘要含有分子动力学模拟的文章数目飞速增长。
尤其是2000年后随着计算能力提升,分子模拟相关研究呈直线上升趋势:
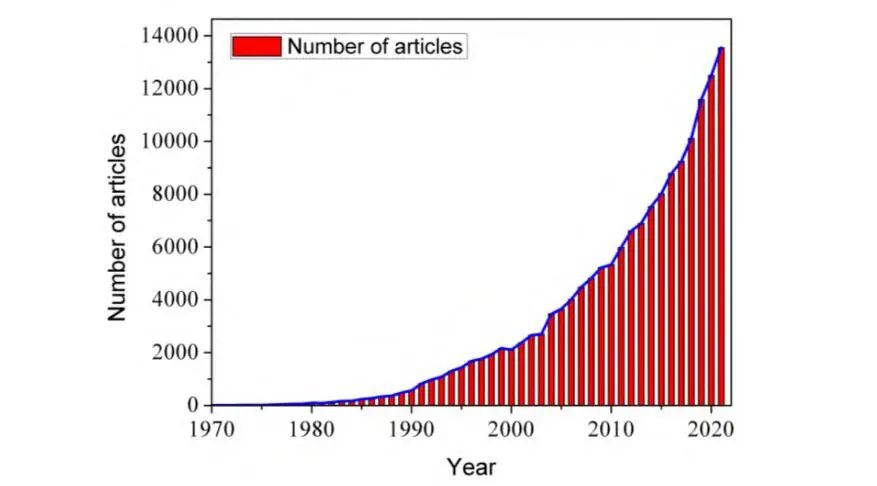
可以说,MD方法在化学及相关研究中的重要地位日趋凸显,但现有计算化学实验多侧重量子化学方法对分子性质的计算。
为普及分子动力学模拟这一有力工具,同时帮助人们理解分子的动态行为,山东大学化学与化工学院苑世领教授团队发表论文。研究者详细列出了模拟步骤,即使是MD小白也能跟着论文内容上手操作。

他们以丙氨酸二肽模型分子为例设计了一个简单的分子动力学模拟实验。通过本实验能初步掌握分子动力学模拟的基本原理、流程和分析方法,同时加深对物理化学中势能面等抽象概念的理解。
☆通讯作者一览:

苑世领,1998年硕士毕业于山东大学化学学院,2003年博士毕业于山大化院,师从江元生院士。主要研究领域和兴趣:
在分子模拟计算方面:1)表面活性剂体系:包括气液界面、体相、固体表面吸附等;2)驱油体系:润湿反转、泡沫驱油、破乳、稠油降粘、PPG驱油等;
3)防污材料体系:贻贝蛋白等在固体表面的吸附等;4)小分子水凝胶体系:Fmoc保护二肽分子、多肽分子等。
在实验方面:小分子水凝胶;石墨烯凝胶体系以及表面活性剂体系。主要论著有:《分子模拟:理论与实验》。
PART-02
MD模拟 分步阐述
一、实验目的及原理
分子动力学模拟主要研究体系中所有粒子的运动状态随时间的演变,在一定的统计力学系综下 通过对相空间进行系综平均(时间平均),获得体系的物理性质和化学性质。从理论上讲,分子动力学模拟才应该算是真正的“计算机实验”。
MD模拟的基本流程包括如下:
1)为所研究问题建立合适的“模型体系”(modeling);
2)确立模型体系的初始状态:包括粒子的坐标参数、速度分布、环境状态等(initialization);
3)为模型体系赋予力场参数(force field);
4)求解牛顿运动方程,直到体系处于平衡状态(equilibrium);
5)从平衡态出发,继续求解运动方程,记录粒子的运动轨迹(production);
6)对平衡后的轨迹进行统计分析(analyze)。
以氨酸模型二肽分子为例,简单说明分子动力学模拟的基本原理和步骤,并初步 探讨温度、溶剂等外部因素对分子构象的影响。
二、实验条件
计算机,Materials Studio软件(Visualizer、Forcite模块)
三、实验内容
1、构建丙氨酸模型二肽分子结构
乙酰基和甲胺基封端的丙氨酸二肽(N-acetyl-N’-methyl-L-alanylamide,NANMA)是一个常用来研 究蛋白质中氨基酸的构象行为的典型模型分子,其分子结构如图2所示。首先采用Visualizer模块 构建其三维结构。
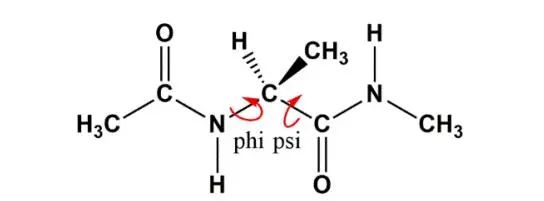
图2:丙氨酸模型二肽结构式
点击菜单栏File-New...在弹出的对话框中选择3D Atomistic。非氢原子完成后单击工具栏H标志完成加氢,再单击其附近的Clean功能调整优化分子结构。完成后可将文件名重命名为NANMA.xsd。
2、分子动力学计算
通常在运行分子动力学计算之前需要对初始模型进行能量极小化(Minimization),即几何优化 (Geometry Optimization),以防止人为搭建的原子模型间有不合理的接触。
在上一步构建分子模型时已采用Visualizer模块的Clean功能进行了初步的构型优化,故可以直接进行分子动力学的模拟。
具体操作如下:打开Forcite计算模块,在Setup标签页将任务设置为Dynamics即动力学计算,再点击右侧More...按钮设置具体模拟参数,如图3所示。
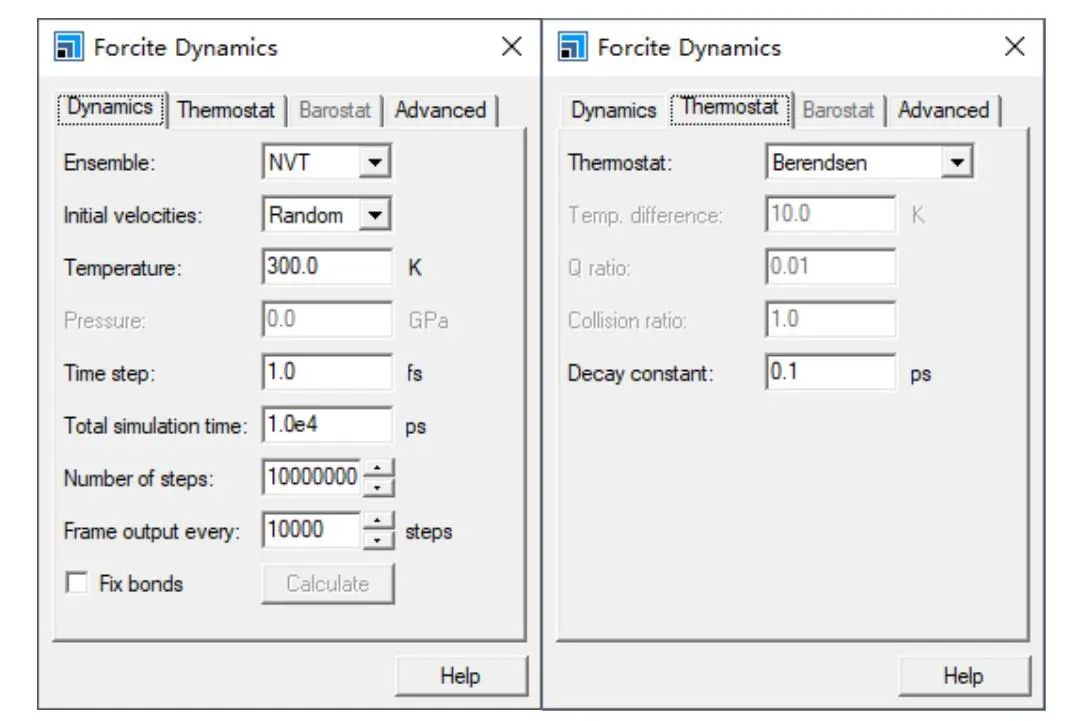
图3:分子动力学计算Setup标签页基本模拟参数设置
在NVT系综下计算(粒子数、温度、体积恒定),随机分配初速度,温度300K,动力学计算积分步长1fs,总共模拟10ns,每间隔10000步记录一次轨迹信息,其中控温算法选择Berendsen方法。
在Energy标签页将力场设置为COMPASS,对于非键相互作用(包括静电相互作用和范德华相互作用)的加和方式均采用Atom based加和法。
Job Control标签页可以设置并行计算的核数和任务描述,应采用尽可能多的CPU核数并行以加速计算,如图4所示。将构型文件NANMA*xsd窗口激活,点击Run即可提交计算,计算进程会在下方任务栏显示。
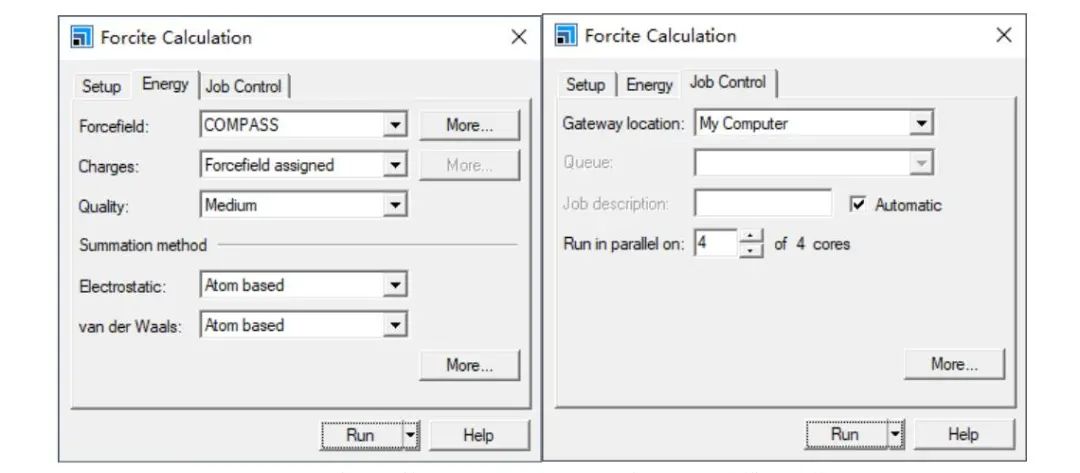
图4:分子动力学计算Energy、Job Control标签页基本模拟参数设置
3、性质分析
1) 动力学轨迹的可视化分析:
动力学计算完成后会生成一条记录原子每一步坐标、速度和受力信息的轨迹文件NANMA.xtd, 通过Animation工具栏可以对体系在动力学计算过程中的变化进行可视化分析,调整好视角之后亦可 以导出为视频文件(File-Export-*.avi)。
2) 骨架二面角随时间的变化和分布:
在获得轨迹的基础上,可以对体系中感兴趣的量进行定量统计分析。
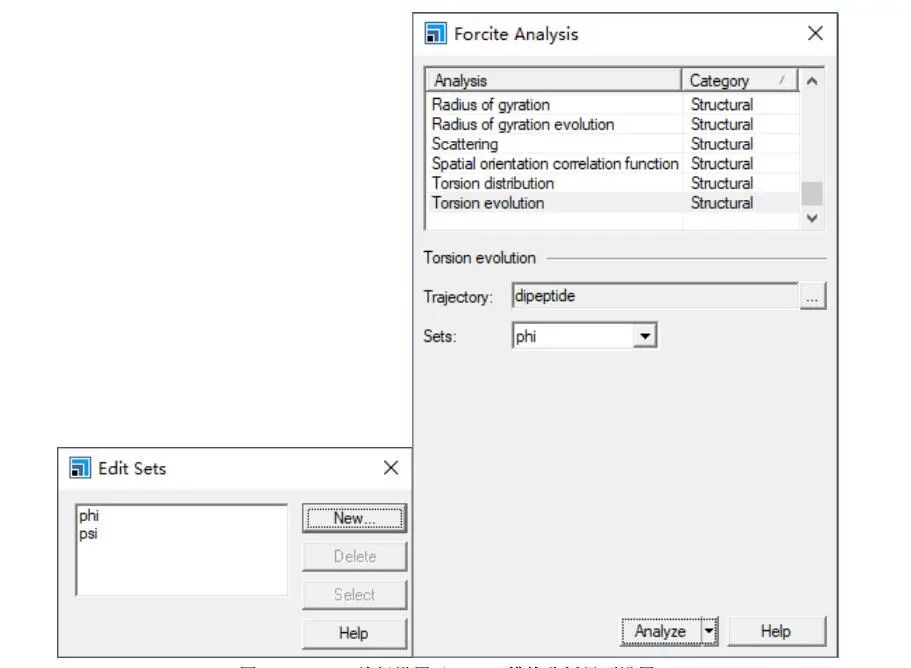
图5:Edit Sets编组设置及Forcite模块分析界面设置
4、高温分子动力学
参考2中的方法可进一步对丙氨酸二肽模型分子在500K温度下的构象变化进行模拟,统计骨架二面角phi和psi随时间的变化,考察其与300K温度下的异同。
显然可以发现在500K温度下,分子的构象变化得更为剧烈,两个二面角的分布也更加宽泛。这也说明升高模拟温度可以帮助分子跨越一些常温条件下难以跨越的势垒,更加充分地对势能面进行采样。
5、淬火动力学
通过对两个温度下进行模拟可发现在低温时分子倾向于在极小点附近振动,在高温时分子则能轻易地跨越势能面上的势垒。
因此可以通过高温分子动力学结合能量极小化的办法找到势能 面上的一批极小点结构(local minimum),在分子动力学中这种方法称为淬火。
具体操作如下:打开Forcite计算模块,在Setup标签页将任务设置为Quench即淬火,再点击右侧More...按钮设置具体淬火参数,每10000步淬火一次;
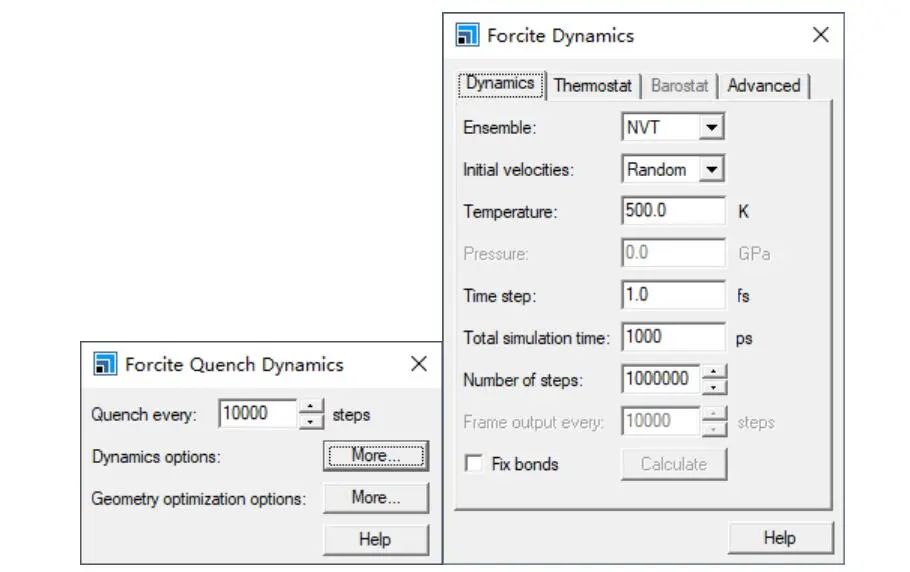
图6:淬火动力学基本模拟参数设置
在NVT系综下计算,随机分配初速度,模拟温度500K,动力学计算积分步长1fs,共模拟1ns,控温算法选择Berendsen方法。
结果文件中*.xtd文件为动力学轨迹文件,* Quench.xtd文件为经能量极小化后的轨迹文件, *Quench.std文件为淬火动力学得到的分子极小点结构和其他信息汇总表。在此基础上可进行一系列的性质分析。
6、模拟退火动力学
通过淬火动力学方法能够找到一批极小点的结构,但是研究者更关心的是分子的全局最小点结构(global minimum),显然在高温动力学的基础上,缓慢地降温,则分子极有可能收敛到该结构。
如果周期性地循环升温-降温则可以找到势能面上的一批极小点结构,这批结构中则有可能存在分子的全局最小点结构。这种周期性升温-降温的方法称作模拟退火动力学方法。
7、团簇结构的分子动力学模拟
采用类似的方法可对丙氨酸二肽模型分子形成的二聚体或与水分子形成的团簇结构进行MD模拟,并考察其组装结构。
注:原子数增多后需要计算的原子对间的相互作用的数目也会增加,导致计算量增加,达到同样的模拟时间的目标下;
为了节约耗时,可以在Setup标签页将模拟步长设置为2fs,同时勾选上Fix bonds,这样在动力学计算过程中程序会约束住与H原子相连的键的振动,可以使用较大的时间步长。
征稿启事
“分子动力学”自创号以来得到了广大科研人士的关注和支持。为更好地服务分子动力学研究和应用,本公众号现长期招聘供稿作者。
真诚欢迎MD相关科研爱好者加入“分子动力学”团队!可发送简历至likeapoem@163.com,邮件主题请注明“姓名+供稿作者”。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









