






参考教程:物种内共线性分析——思路以及踩坑总结(二)-CSDN博客
首先下载所需物种参考蛋白组序列,建议选择ensemble plant 或者 Phytozome数据库
#具体文件具体分析 gff文件筛选
$grep '\smRNA\s' EVM.final.gene.gff3 | awk '{print $1"\t"$4"\t"$5"\t"$9}' |awk -F 'ID=' '{print $1$2}'|awk -F 'Parent=' '{print $1}'|awk '{print $1"\t"$4"\t"$2"\t"$3}' >pde.gff
$grep '\smRNA\s' Ptrichocarpa_444_v3.1.gene.gff3 | awk '{print $1"\t"$4"\t"$5"\t"$9}'|awk -F '.v3.1;' '{print $1}'|awk -F 'ID=' '{print $1$2}'|awk '{print $1"\t"$4"\t"$2"\t"$3}' > ptr.gff编写适用于物种内共线性分析的脚本,使用时仅需要修改
#A script for inner_blast and collinear analysis
#提前下载diamond 和 MCScanX工具
#input需要为绝对路径
input_pep=/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/peptide/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.pep.all.fa
#output为前缀名
output=inner_maize
#gff为绝对路径
gff=/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/filtered_gff/zm.gff
#/home/xintong/gene_family_analysis/gene_fami_ana/inner_species
diamond makedb --in $input_pep -d $output
diamond blastp -d ${output}.dmnd -q $input_pep -o ${output}.blast --sensitive
cp $gff ${output}.gff
MCScanX $output
--sensitive 该模式适合比对较长的序列,默认模式适合比对short reads
-e 阈值evalue的设定,模式是0.001,也可以继续降低阈值
若需要多物种共线性分析-不考虑种间,则参考以下脚本;若多物种间共线性分析-同时考虑种内和种间,则把所有物种的蛋白组文件合并为一个新的fa文件,当做种内共线性处理,见上一个脚本。
#A script for inter_species blast and collinear
#提前下载diamond 和 MCScanX工具
#以下四项需要为绝对路径 分别代表query和subject的pep.fa文件和filtered.gff文件
query_pep=
subject_pep=
#output为前缀名
output=
#database为subject_database的前缀名
database=
#以subject_pep为基础构建blast database 输出前缀为output
diamond makedb --in $subject_pep -d $database
#使用output.dmnd,用query_pep去搜索,输出blast文件为output.blast,参数为sensitive
diamond blastp -d ${database}.dmnd -q $query_pep -o ${output}.blast --sensitive
#连续实行多次此脚本后,实现多物种两两间相互blast,合并所有的blast文件和gff文件,并保持相同的前缀,参考MCScanX用法,进行共线性分析
#玉米pep.fa和filtered.gff存储地址
/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/peptide/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.pep.all.fa
/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/filtered_gff/zm.gff#大豆pep.fa和filtered_gff存放地址
/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/peptide/Glycine_max.Glycine_max_v2.1.pep.all.fa
/home/xintong/gene_family_analysis/gene_fami_ana/origin_genome/filtered_gff/Gm_filtered.gff3接下来将.collinearity和gff文件下载到本地,使用TBtools进行分析
1. Text Merge for MCScanX模块,模式选择collinearity
将collinearity文件转化为txt文件,去除多余信息
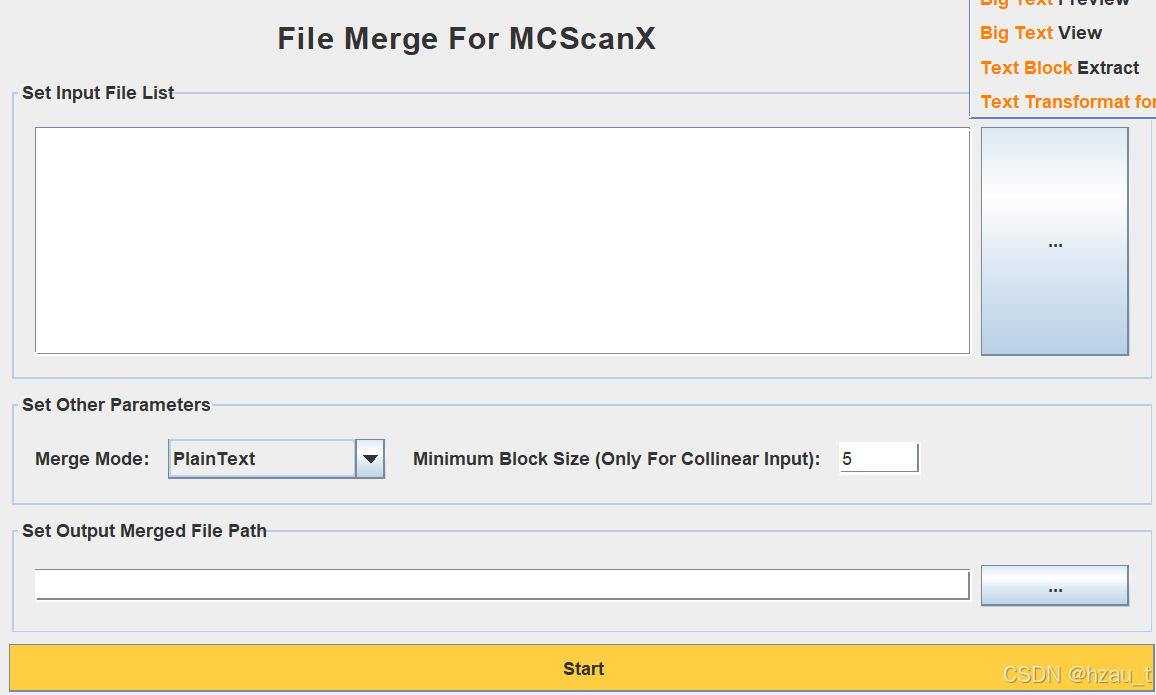
2. Text block Extract
过滤PLATZ家族的共线性关系
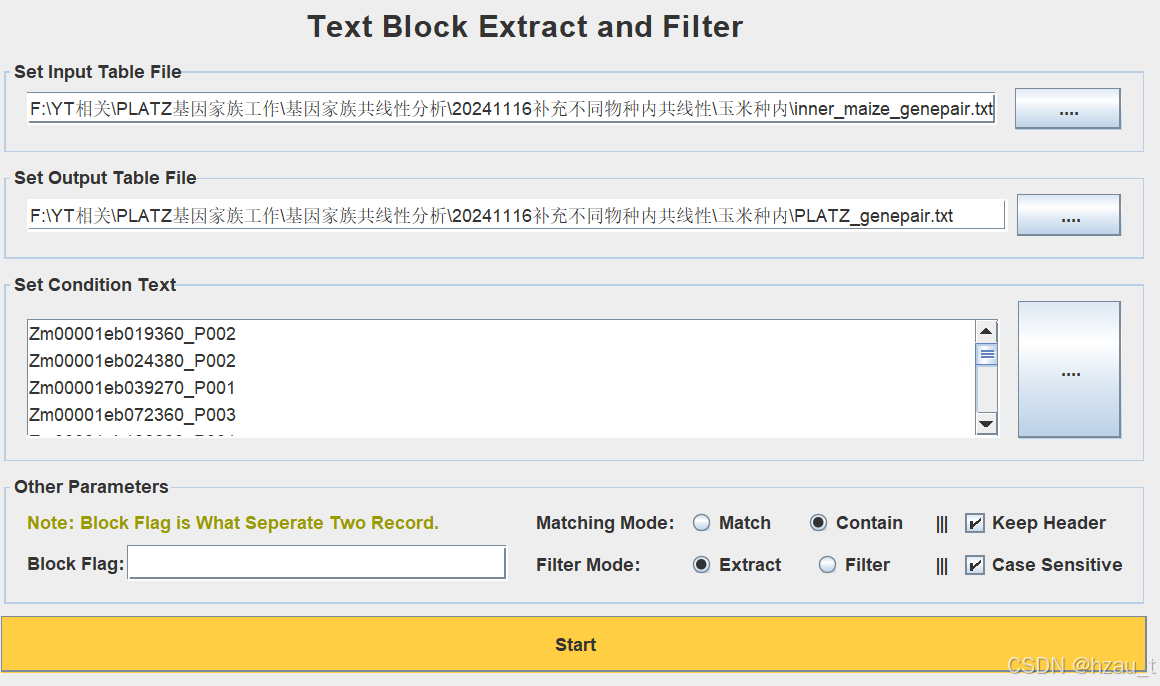
3. Text Transformation for micro-Synteny view
借助PLATZ家族成员的共线性关系以及过滤后的gff文件获得link-location关系
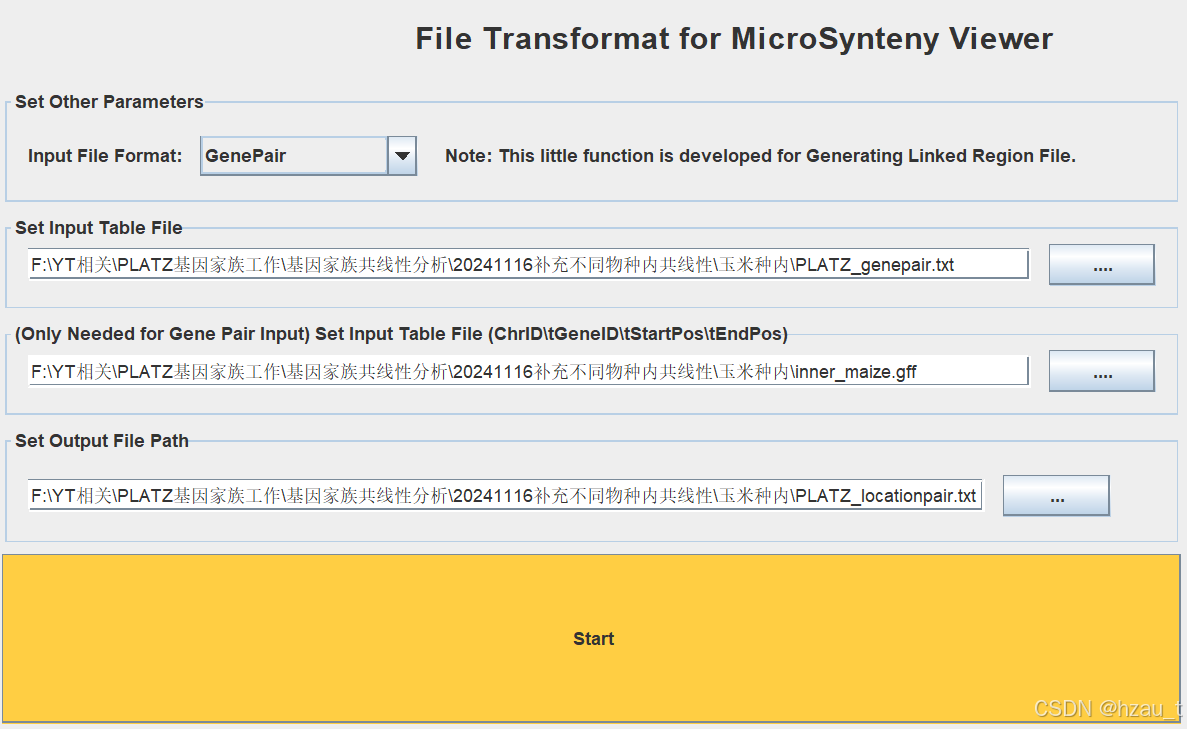
处理过程中发现inner_maize.gff文件不对,第一列以mz为前缀,需要替换为Zm为前缀,以适应maize.size文件,代码如下:
sed -i 's/mz/Zm/' inner_maize.gff这里的-i选项表示直接在原文件上进行修改,s表示替换操作,mz是要被替换的内容,zm是替换后的内容,g表示全局替换(即每一行中所有匹配的都替换,如果不加g,则每行只替换第一次出现的情况)。
1)GC含量/GCskew/未知碱基
核酸组成是基因组的一个基本特征。例如,GC含量与编码基因和功能DNA元素的密度相关;GC-skew是一种度量鸟嘌呤和胞嘧啶链偏好的指数,可以帮助检测细菌环形染色体中的DNA复制起始位点。未知碱基(N)分布可从一定程度上显示基因组组装质量。TBtools中的“FastaWindowStat”功能可用于快速将基因组序列文件中的GC含量、GC偏斜和N比率制成表格。
功能位置:“SequenceToolkit”->“FastaTools”->“FastaWindowStat”
输入基因组序列文件:aliana.genome.fna
输出文件前缀:genome.Window.Stat
将生成三个前缀相同的文件。他们是
genome.Window.Stat.GCratio”
“genome.Window.Stat.GCskew”
“genome.Window.Stat.Nratio”
之后可以在基因组骨架上添加这些信息的轨道。如下
2)基因密度
基因组范围内的基因密度分布通常通过Circos图进行可视化。TBtools有一个方便的功能“GeneDensityProfile”,从基因结构注释文件中计算基因密度,通常为GFF3和GTF格式。
功能位置:“SequenceToolkits”->“GFF3/GTFManipulate”->“GeneDensityProfile”
输入文件:genome.gff3
输出文件:GeneDensity.profile
在“ControlDialog”面板中,单击“Add”以获得附加轨道,拖放基因密度信息文件(“GeneDensity.profile”),选择热图模式,然后刷新绘图。如果基因密度分布明显有偏差,可以在左下角面板的“HeatColorScale”菜单中尝试不同的颜色缩放模式,以获得更好的视图。
sed -i 's/^/Zm/' maizegendensity
for file in ./maize.*; do
echo $file
if [ -f "$file" ]; then
sed -i 's/^/Zm/' "$file"
fi
done


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









